📋 Key Information Summary
- Behçet syndrome is a systemic vasculitis characterised by recurrent oral and genital ulcers, ocular inflammation, and variable involvement of vessels, joints,神经系统, and gastrointestinal tract.
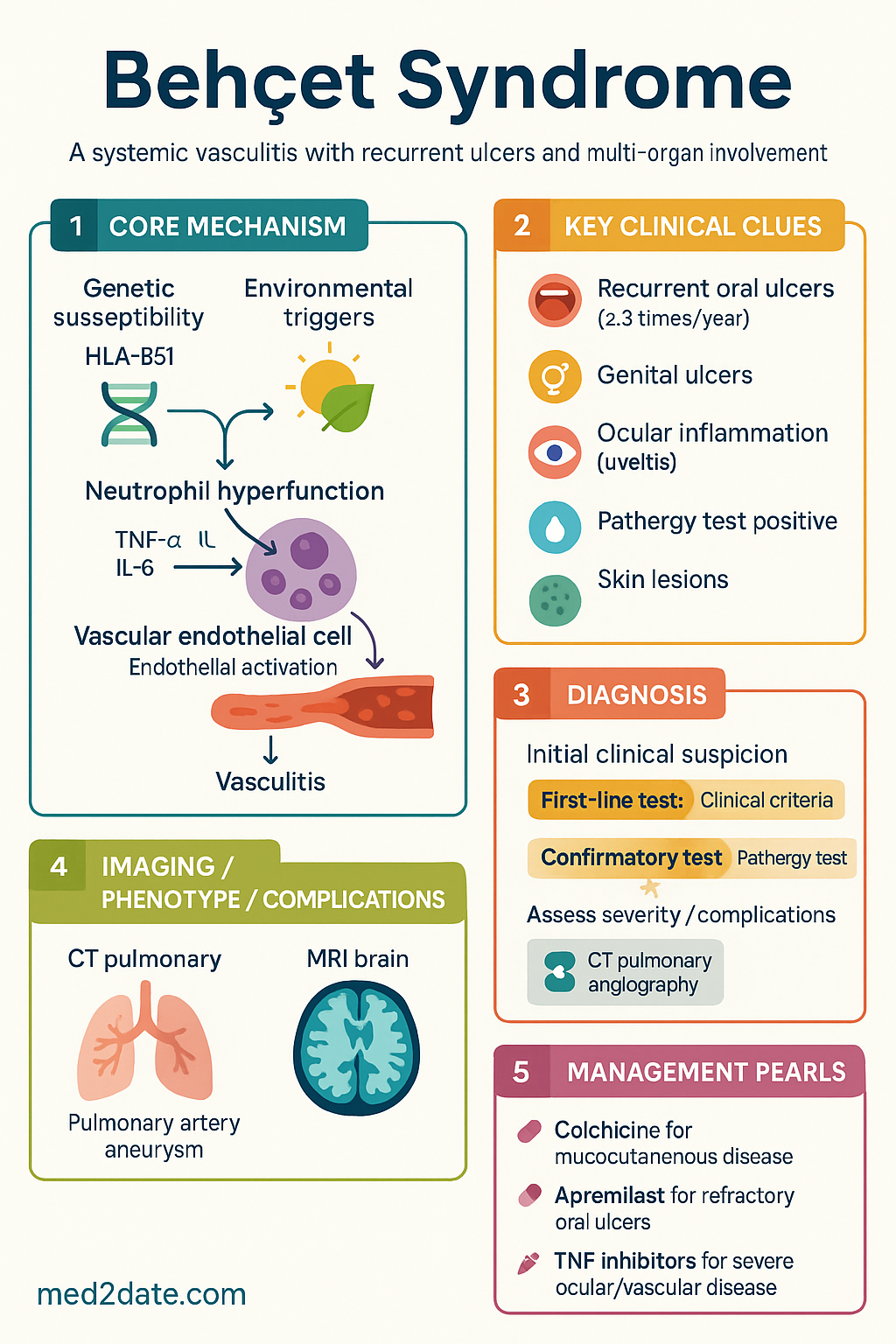
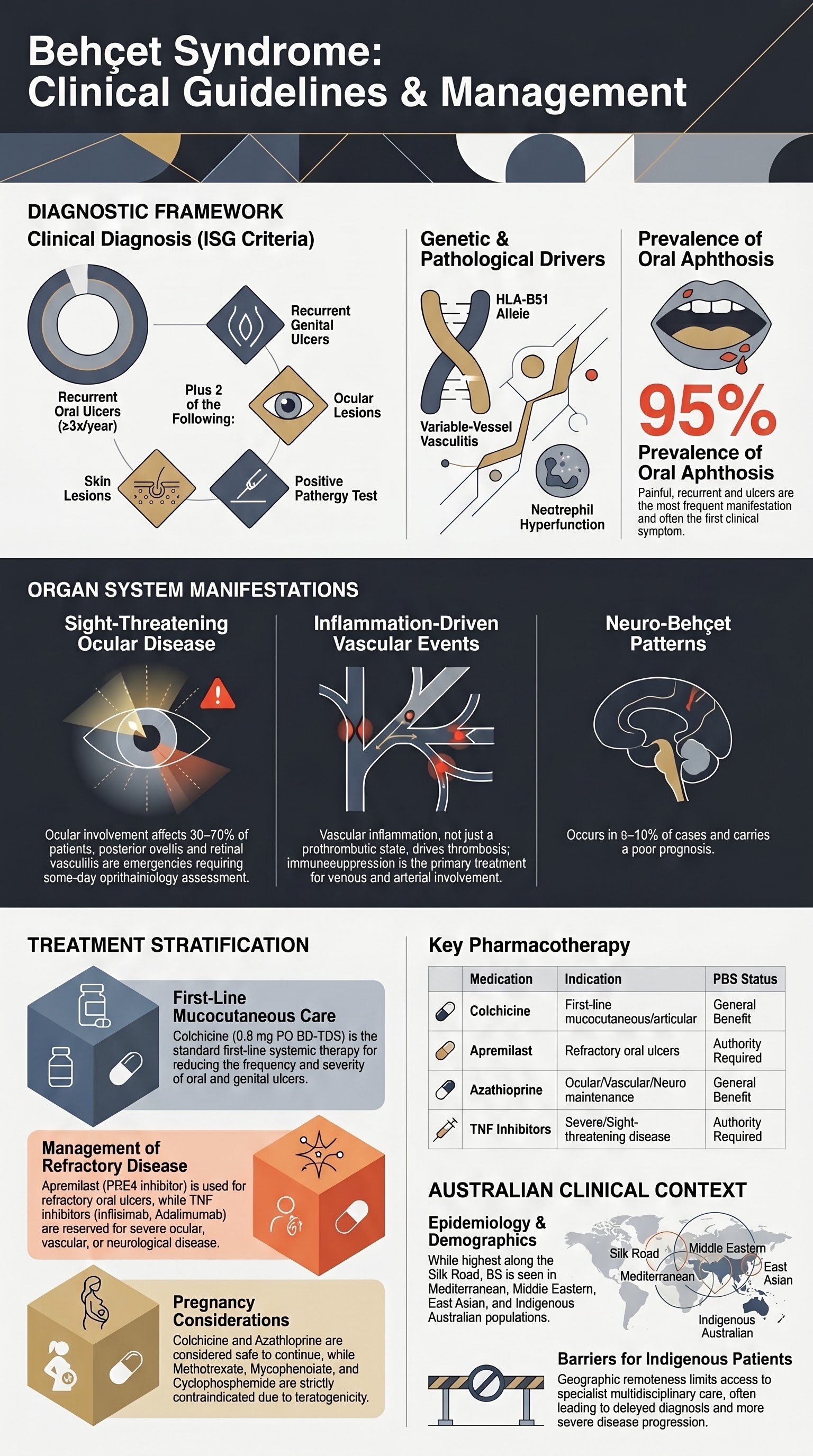
- Diagnosis is clinical; International Study Group criteria require recurrent oral ulcers (≥3 times/year) plus two of: recurrent genital ulcers, ocular lesions, skin lesions, or a positive pathergy test.
- Ocular disease (anterior/posterior uveitis, retinal vasculitis) is sight-threatening and requires urgent ophthalmology referral and aggressive immunosuppression.
- Major vessel involvement, especially pulmonary artery aneurysm, carries high mortality; CT pulmonary angiography is key for diagnosis.
- Neuro-Behçet can present with parenchymal involvement (brainstem) or cerebral venous thrombosis; MRI brain with contrast is essential.
- First-line therapy for mucocutaneous disease is colchicine. Apremilast is PBS-listed for oral ulcers refractory to colchicine.
- For severe ocular, vascular, or neurological disease, conventional immunosuppressants (azathioprine, cyclosporine, mycophenolate) and/or TNF inhibitors (infliximab, adalimumab) are first-line biologics.
- Anticoagulation for vascular thrombosis is controversial; immunosuppression is the primary treatment for inflammation-induced thrombosis.
- Disease activity monitoring uses the Behçet's Disease Current Activity Form (BDCAF).
- Considerations for Aboriginal and Torres Strait Islander patients include access to specialist care, potential for more severe disease, and culturally safe management.
Introduction & Australian Epidemiology
Behçet syndrome (BS) is a chronic, relapsing systemic inflammatory disorder classified among the variable-vessel vasculitides. It is characterised by a triad of recurrent oral aphthosis, genital ulceration, and ocular inflammation, but can affect virtually any organ system. The pathogenesis involves a complex interplay between genetic susceptibility (strongly associated with HLA-B51) and environmental triggers, leading to neutrophil hyperfunction and vasculitis.
In Australia, BS is considered rare, with prevalence highest in populations along the ancient Silk Road (Middle East, East Asia). However, due to migration, it is encountered in Australian rheumatology, ophthalmology, and dermatology practice. There is a paucity of Australian-specific epidemiological data, but it is observed in patients of Mediterranean, Middle Eastern, and East Asian descent, as well as in Indigenous Australians. The male-to-female ratio is approximately 1:1 in Australia, though males often have more severe disease.
Recurrent Oral Ulcers
Recurrent oral aphthous ulcers are the hallmark and most frequent manifestation (≈95% of patients), often the first symptom. They are painful, round or oval, with a central yellow-grey necrotic base surrounded by a erythematous halo. Three types occur: minor (most common, <10 mm), major (>10 mm, deep, may scar), and herpetiform (clustered small ulcers).
Management is stepped:
- Topical: High-potency topical corticosteroids (e.g., clobetasol propionate 0.05% gel/orabase) for individual ulcers.
- First-line systemic: Colchicine 0.5 mg PO BD-TDS (max 2 mg/day). Reduces frequency and severity.
- Second-line systemic for refractory ulcers: Apremilast 30 mg PO BD (PBS Authority Required). An oral PDE4 inhibitor; requires authority approval for Behçet's disease with severe oral ulcers.
Genital Ulcers
Genital ulcers occur in ~80% of patients and are morphologically similar to oral ulcers but tend to be larger, deeper, and more frequently leave scars. In males, they commonly affect the scrotum and shaft of the penis. In females, they occur on the vulva and vagina. They are often a major cause of morbidity due to pain and scarring.
Management mirrors oral ulcer therapy. Topical agents (potent corticosteroids, sucralfate paste) are used for symptomatic relief. Systemic therapy with colchicine is first-line. Apremilast and other systemic agents used for severe mucocutaneous disease (e.g., dapsone, azathioprine) are also effective. Recurrent scarring may require surgical consultation.
Ocular Disease (Uveitis, Retinal Vasculitis)
Ocular involvement affects 30–70% of patients and is a major cause of morbidity. It is often bilateral and recurrent. Manifestations include:
- Anterior uveitis: Pain, redness, photophobia.
- Posterior uveitis & retinal vasculitis: Floaters, blurred vision. Can lead to retinal ischaemia, neovascularisation, vitreous haemorrhage, and blindness.
- Panuveitis: Combined inflammation.
Treatment: Requires combined ophthalmology-rheumatology management.
- Acute flares: High-dose systemic corticosteroids (e.g., IV methylprednisolone 1 g/day for 3 days, then oral prednisolone 1 mg/kg/day).
- Maintenance: Azathioprine (2–3 mg/kg/day) is often first-line steroid-sparing agent. Mycophenolate mofetil (1–1.5 g BD) is an alternative.
- Refractory/severe disease: TNF inhibitors are first-line biologics. Infliximab (5 mg/kg IV at 0, 2, 6 weeks then 6–8 weekly) or Adalimumab (40 mg SC every 1–2 weeks) are PBS Authority options.
- Local therapy: Periocular/intravitreal corticosteroids (e.g., dexamethasone implant) may be used.
Vascular Involvement
Venous thrombosis is more common than arterial. Superficial thrombophlebitis and deep vein thrombosis (DVT) are frequent. Pulmonary artery aneurysms (PAA) are rare but carry a high mortality rate due to rupture and haemoptysis.
Investigations: CT pulmonary angiography (CTPA) for suspected PAA; Doppler ultrasound for DVT.
Treatment:
- Venous thrombosis: Systemic corticosteroids + immunosuppressants (cyclophosphamide for severe, azathioprine for maintenance). Anticoagulation is often added for 6–12 months but is debated; risk-benefit must be individualised (concurrent PAA risk).
- Pulmonary artery aneurysm: High-dose corticosteroids + cyclophosphamide pulse therapy. TNF inhibitors (infliximab) are highly effective. Surgery/embolisation is reserved for life-threatening haemorrhage.
Neurological Involvement
Neuro-Behçet occurs in 5–10% and is a poor prognostic factor. Two main patterns:
- Parenchymal: Most common. Brainstem/meningeal involvement, hemiparesis, cranial nerve palsies, psychiatric symptoms. MRI shows T2/FLAIR hyperintensities in brainstem, basal ganglia, thalamus.
- Non-parenchymal: Cerebral venous thrombosis (CVT), intracranial hypertension.
Investigations: Urgent MRI brain with contrast and MR venography. Lumbar puncture may show neutrophilic pleocytosis.
Treatment:
- Acute parenchymal: IV methylprednisolone (1 g/day x 3–5 days) followed by oral taper.
- Maintenance: Azathioprine, mycophenolate, or methotrexate.
- Refractory: TNF inhibitors (infliximab preferred) or interferon-alpha.
- CVT: Anticoagulation + corticosteroids.
Treatment Overview (Colchicine, Apremilast, Biologics)
Treatment is stratified by organ involvement severity. No single agent is curative; therapy aims to suppress inflammation and prevent damage.
Key Medications
Special Populations
📚 References
- 1. International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-347.
- 2. Yazici Y, Hatemi G, Bodaghi B, et al. Behçet syndrome. Nat Rev Dis Primers. 2021;7(1):67.
- 3. Saadoun D, Wechsler B. Behçet's disease. Orphanet J Rare Dis. 2012;7:20.
- 4. Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behçet's syndrome. Ann Rheum Dis. 2018;77(6):808-818.
- 5. Levy-Clarke G, Jabs DA, Read RW, et al. Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology. 2014;121(3):785-796.e3.
- 6. Mesbah R, Remmers EF, Oruc O, et al. Genetic susceptibility to Behçet's disease: role of genes involved in the IL-23/Th17 pathway. Curr Opin Rheumatol. 2021;33(1):67-74.
- 7. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2023 summary report. Canberra: AIHW; 2023.
- 8. The Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice. 10th edn. East Melbourne, Vic: RACGP; 2024.
- 9. National Health and Medical Research Council (NHMRC). Australian guidelines for the clinical care of people with COVID-19. Canberra: NHMRC; 2023. (Referenced for immunosuppression principles).
- 10. Alibaz-Oner F, Sawalha AH, Direskeneli H. Management of Behçet's disease. Curr Opin Rheumatol. 2022;34(1):30-36.
- 11. Zeidan MJ, Saadoun D, Garrido M, et al. Behçet's disease physiopathology: a contemporary review. Auto Immun Highlights. 2016;7(1):4.