📋 Key Information Summary
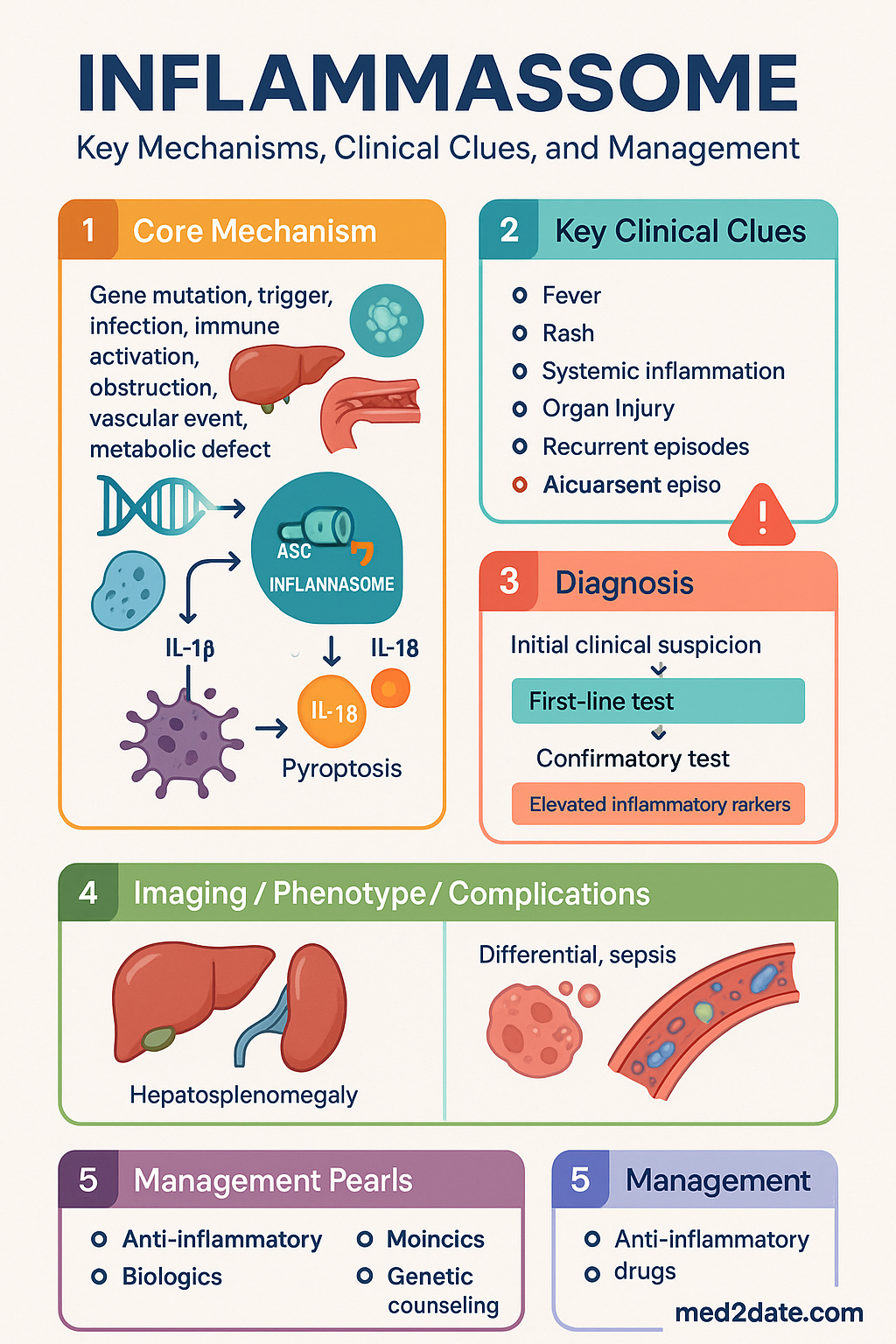
- Inflammasomes are multiprotein complexes that activate caspase-1, leading to cleavage and secretion of IL-1β and IL-18 and induction of pyroptosis.
- The NLRP3 inflammasome is the most clinically significant and best-characterised member, linked to a spectrum of autoinflammatory diseases.
- NLRP3 activation requires two signals: priming (NF-κB-mediated transcription of pro-IL-1β and NLRP3) and activation (NLRP3 oligomerisation triggered by diverse stimuli).
- NLRP3-associated diseases include cryopyrin-associated periodic syndromes (CAPS), familial Mediterranean fever (FMF), gout, type 2 diabetes, Alzheimer disease, and atherosclerosis.
- Gain-of-function NLRP3 mutations cause CAPS (Muckle–Wells syndrome, familial cold autoinflammatory syndrome, NOMID/CINCA) with continuous IL-1β production.
- IL-1-targeted therapies — anakinra, canakinumab, and rilonacept — are PBS-listed for specific autoinflammatory indications in Australia.
- Colchicine remains first-line for FMF and acute gout by inhibiting NLRP3 inflammasome assembly and neutrophil chemotaxis.
- The CANTOS trial demonstrated cardiovascular benefit of IL-1β blockade with canakinumab, establishing the inflammasome–cardiovascular disease axis.
- Inflammasome dysregulation contributes to metabolic syndrome, neurodegeneration, and chronic kidney disease — emerging therapeutic frontiers.
- Aboriginal and Torres Strait Islander peoples experience higher rates of gout and chronic inflammatory diseases; equitable access to biologic therapies must be ensured.
- Diagnosis requires integration of clinical phenotype, inflammatory biomarkers (CRP, SAA, ESR), genetic testing for monogenic autoinflammatory diseases, and exclusion of infection/autoimmunity.
- Ongoing research targets NLRP3-specific inhibitors (e.g., MCC950/CRID3, dapansutrile) and gasdermin D inhibitors as future therapeutic strategies.
Introduction & Australian Context
The inflammasome is a high-molecular-weight multiprotein complex that serves as a molecular platform for the activation of pro-inflammatory caspases, principally caspase-1. Upon assembly, active caspase-1 proteolytically cleaves the precursor cytokines pro-interleukin-1β (pro-IL-1β) and pro-interleukin-18 (pro-IL-18) into their biologically active forms, which are then secreted to drive local and systemic inflammation. Caspase-1 also cleaves gasdermin D (GSDMD), whose N-terminal fragment forms membrane pores — a process termed pyroptosis — resulting in inflammatory cell death and further release of intracellular inflammatory mediators.
The inflammasome concept was first described by Tschopp and colleagues in 2002 with the characterisation of the NLRP1 inflammasome. Since then, multiple inflammasome sensors have been identified, including NLRP3, NLRC4, AIM2, pyrin, and NLRP6. Of these, the NLRP3 inflammasome has received the most intensive investigation owing to its central role in both monogenic autoinflammatory disorders and common multifactorial diseases such as gout, atherosclerosis, type 2 diabetes mellitus, and neurodegenerative conditions.
In Australia, the clinical relevance of inflammasome biology extends across multiple specialties. Autoinflammatory diseases, though individually rare, collectively affect approximately 1 in 1,000–5,000 persons, with increasing recognition due to improved genetic diagnostics available through services such as the Australian Genomics Health Alliance. Gout — now understood as an NLRP3-mediated IL-1β-driven disease — affects approximately 5.2% of Australian men and 1.7% of women, with markedly higher prevalence among Māori, Pacific Islander, and Aboriginal and Torres Strait Islander communities. The expanding therapeutic armamentarium targeting IL-1 cytokines and inflammasome components has created a need for Australian clinicians to understand the underlying biology, recognise autoinflammatory phenotypes, and apply evidence-based treatment strategies aligned with PBS criteria and local antimicrobial stewardship principles.
NLRP3 Inflammasome
Structure and Components
The NLRP3 inflammasome is composed of three core components: the sensor protein NLRP3 (nucleotide-binding oligomerisation domain, leucine-rich repeat and pyrin domain-containing 3, also termed cryopyrin), the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and the effector protease pro-caspase-1. Upon activation, NLRP3 undergoes oligomerisation through its NACHT domain, recruiting ASC via homotypic pyrin domain (PYD–PYD) interactions. ASC in turn polymerises into large filamentous structures called ASC specks, which recruit pro-caspase-1 through CARD–CARD interactions, enabling proximity-induced auto-proteolytic activation of caspase-1.
The NLRP3 gene (CIAS1) is located on chromosome 1q44 and encodes a 1,036-amino-acid protein. NLRP3 expression is largely restricted to cells of the myeloid lineage, including monocytes, macrophages, dendritic cells, and neutrophils, though expression can be induced in epithelial and endothelial cells under inflammatory conditions.
Key Inflammasome Family Members
| Inflammasome | Sensor Protein | Activating Stimuli | Associated Diseases |
|---|---|---|---|
| NLRP3 | Cryopyrin | ATP, crystals (MSU, CPPD, silica), K⁺ efflux, ROS, lysosomal disruption, viral RNA | CAPS, gout, pseudogout, atherosclerosis, T2DM, Alzheimer disease |
| NLRP1 | NLRP1 / CARD8 | Toxoplasma, UV radiation, muramyl dipeptide, anthrax lethal toxin | NLRP1-associated autoinflammatory disease with skin and airway inflammation |
| NLRC4 | NLRC4 (IPAF) | Flagellin, rod proteins (Salmonella, Legionella, Shigella) | NLRC4-MAS, NLRC4 autoinflammatory disease |
| AIM2 | Absent in melanoma 2 | Cytosolic dsDNA (bacterial, viral, self) | Psoriasis, lupus-like, colorectal cancer |
| Pyrin | MEFV | RhoA inactivation by bacterial toxins (C. difficile TcdB, Burkholderia) | Familial Mediterranean fever (FMF) |
NLRP3 Gain-of-Function Mutations and CAPS
Cryopyrin-associated periodic syndromes (CAPS) are autosomal dominant disorders caused by gain-of-function mutations in CIAS1/NLRP3, resulting in constitutive inflammasome activation and uncontrolled IL-1β production. CAPS represents a phenotypic spectrum:
Activation Mechanisms
Two-Signal Model
Canonical NLRP3 inflammasome activation requires two sequential signals. This two-step mechanism ensures that inflammasome assembly occurs only in the context of appropriate immune stimulation, preventing inappropriate activation by endogenous danger signals.
Non-Canonical Inflammasome Activation
Non-canonical inflammasome activation involves caspase-11 (murine) / caspase-4 and caspase-5 (human), which directly bind cytosolic lipopolysaccharide (LPS) from Gram-negative bacteria. Activated caspase-4/5/11 cleave gasdermin D, inducing pyroptosis, and trigger NLRP3-dependent caspase-1 activation for IL-1β/IL-18 maturation. This pathway is critical for defence against cytosolic-invasive bacteria (Salmonella, Legionella, Citrobacter) and underlies the lethal endotoxaemia observed in sepsis.
Alternative Inflammasome Activation
An alternative NLRP3 activation pathway has been described in human monocytes stimulated with LPS alone (without ATP or other second signals), involving TLR4–TRIF–RIPK1–FADD–caspase-8 signalling. This pathway produces IL-1β without pyroptosis and may be relevant in sterile inflammatory conditions such as transfusion-related acute lung injury (TRALI) and gout flares.
Inflammatory Diseases
Monogenic Autoinflammatory Diseases
Monogenic autoinflammatory diseases are driven by mutations in innate immune genes that lead to uncontrolled inflammasome activation in the absence of autoantibodies or autoreactive T cells. They typically present in childhood with recurrent fevers and systemic inflammation.
| Disease | Gene | Inflammasome | Key Features | 1st-Line Rx |
|---|---|---|---|---|
| FMF | MEFV (Pyrin) | Pyrin | 1–3 day episodes of fever, serositis, erysipelas-like erythema; AA amyloidosis | Colchicine (PBS listed) |
| TRAPS | TNFRSF1A | Multiple (enhanced NF-κB) | Prolonged episodes (1–3 wk), migratory rash, periorbital oedema, myalgia | IL-1 blockade (anakinra/canakinumab) |
| CAPS | NLRP3 (CIAS1) | NLRP3 | See FCAS/MWS/NOMID spectrum above | IL-1 blockade |
| MKD/HIDS | MVK | NLRP3 (mevalonate pathway) | Recurrent fever, lymphadenopathy, abdominal pain, rash triggered by vaccination | IL-1 blockade / corticosteroids |
| Blau syndrome | NOD2/CARD15 | NOD2-dependent | Granulomatous uveitis, arthritis, skin rash (early childhood onset) | Methotrexate + corticosteroids |
Common Inflammasome-Associated Diseases
Beyond monogenic disorders, NLRP3 inflammasome activation is a key driver in several prevalent multifactorial diseases relevant to Australian primary care:
Gout
Monosodium urate (MSU) crystal deposition in joints and periarticular tissues activates the NLRP3 inflammasome via lysosomal disruption and cathepsin B release, producing IL-1β-driven acute inflammation. Gout prevalence in Australia is approximately 5.2% in men and 1.7% in women, with significantly higher rates in Aboriginal and Torres Strait Islander peoples, Māori, and Pacific Islander populations. Colchicine inhibits NLRP3 inflammasome assembly by blocking microtubule-dependent ASC speck formation and neutrophil recruitment.
Atherosclerosis and Cardiovascular Disease
Cholesterol crystals in atherosclerotic plaques activate NLRP3, driving chronic vascular inflammation. The landmark CANTOS trial demonstrated that canakinumab (anti-IL-1β monoclonal antibody) reduced major adverse cardiovascular events by 15% in patients with prior myocardial infarction and elevated hs-CRP (≥ 2 mg/L), providing proof-of-concept for the inflammatory hypothesis of atherosclerosis.
Type 2 Diabetes Mellitus and Metabolic Syndrome
Saturated fatty acids, ceramides, and hyperglycaemia activate NLRP3 in adipose tissue macrophages, contributing to insulin resistance and β-cell dysfunction. IL-1β-mediated β-cell apoptosis has been demonstrated in vitro and in human islets. Clinical trials of anakinra in T2DM have shown improved glycaemic control and β-cell function.
Neurodegenerative Diseases
Microglial NLRP3 activation by β-amyloid and α-synuclein aggregates contributes to neuroinflammation in Alzheimer disease and Parkinson disease respectively. ASC specks released from microglia can seed further amyloid aggregation, creating a pathological amplification loop. Genetic studies show NLRP3 loss-of-function variants are associated with reduced Alzheimer disease risk.
Chronic Kidney Disease
NLRP3 activation in renal tubular epithelial cells and infiltrating macrophages by uraemic toxins, calcium phosphate crystals, and oxalate promotes tubulointerstitial fibrosis. IL-1β and IL-18 amplify renal inflammation and contribute to progression of diabetic nephropathy and IgA nephropathy.
Clinical Presentation & Diagnostic Approach
Suspect an inflammasome-mediated autoinflammatory disease in patients with:
- Recurrent, stereotyped episodes of fever without infectious cause
- Elevated acute-phase reactants (CRP, SAA, ESR) between episodes or at baseline
- Characteristic rash (urticaria-like in CAPS, erysipelas-like in FMF, migratory erythema in TRAPS)
- Serositis (pleuritis, peritonitis, pericarditis)
- Family history of recurrent fevers or amyloidosis
- Symptom onset in infancy or early childhood
Diagnostic workup:
- Inflammatory markers: CRP, SAA, ESR, FBC, ferritin (to exclude macrophage activation syndrome)
- Genetic testing: Targeted gene panel or whole-exome sequencing (available via Australian Genomics, clinical genetics services)
- Exclude infection, malignancy, and autoimmune disease (ANA, ENA, ANCA where appropriate)
- Consider serum immunoglobulin D (IgD) for MKD/HIDS screening
- Joint aspiration with polarised light microscopy for crystal arthropathy (gout, pseudogout)
Investigations
Therapeutic Targets
Current Approved Therapies
Emerging and Investigational Therapies
| Agent | Target | Mechanism | Development Stage |
|---|---|---|---|
| MCC950 / CRID3 | NLRP3 NACHT domain | Directly blocks NLRP3 oligomerisation and ATPase activity | Phase I/II (discontinued for hepatotoxicity concerns; modified analogues in development) |
| Dapansutrile (OLT1177) | NLRP3 ATPase | Oral NLRP3 inhibitor; prevents inflammasome assembly | Phase II — heart failure with reduced EF; gout |
| NT-0796 | NLRP3 (brain-penetrant) | CNS-penetrant NLRP3 inhibitor for neuroinflammation | Phase I — neurodegenerative diseases |
| Nirsevimab / anti-NEK7 | NEK7–NLRP3 interaction | Disrupts NEK7-mediated NLRP3 activation downstream of K⁺ efflux | Preclinical |
| Dimethyl fumarate | NRF2 / GSDMD | Activates NRF2 antioxidant pathway; directly modifies GSDMD Cys191 to block pyroptosis | Approved (MS); repurposing for autoinflammation |
| Disulfiram | GSDMD | Covalently modifies GSDMD Cys191, preventing pore formation and pyroptosis | Phase II — COVID-19, alcohol use disorder repurposing |
| Tranilast | NLRP3 NACHT domain | Binds NACHT domain, preventing NLRP3 oligomerisation; also inhibits ASC speck formation | Preclinical (approved as anti-allergic agent in Japan/Korea) |
Therapeutic Algorithm for Autoinflammatory Disease
Monitoring on IL-1-Targeted Therapy
- Efficacy: Symptom diary, CRP/SAA at 1 month then 3-monthly; target SAA < 10 mg/L in CAPS to prevent amyloidosis
- Safety: FBC at 1, 3, 6 months then 6-monthly (neutropenia risk); LFTs; screen for latent TB (QuantiFERON/TST) before commencing
- Infections: Active infection is a relative contraindication; counsel on infection precautions; avoid live vaccines
- Pregnancy: Anakinra is Category B1 (limited human data, no teratogenicity in animal studies); canakinumab is Category B1. Discuss risks/benefits in pre-conception counselling. Colchicine is Category D — however, low-dose continuation in FMF is recommended (risk of flare outweighs teratogenic risk).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10(2):417–426.
- 2. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489.
- 3. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015;10:395–424.
- 4. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease (CANTOS). N Engl J Med. 2017;377(12):1119–1131.
- 5. Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76(6):942–947.
- 6. Shohat M, Halpern GJ. Familial Mediterranean fever — a review. Genet Med. 2011;13(6):487–498.
- 7. Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388(10055):2039–2052.
- 8. AIHW (Australian Institute of Health and Welfare). Gout and its comorbidities. Bulletin 147. Canberra: AIHW; 2018.
- 9. Liew FY, McInnes IB. The role of innate mediators in inflammatory response. Mol Immunol. 2002;38(12–13):887–890.
- 10. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021.
- 11. Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255.
- 12. Savic S, Dickie LJ, Wittmann M, McDermott MF. Autoinflammatory syndromes and cellular stress responses. Immunology. 2012;136(1):1–12.
- 13. RACGP. Musculoskeletal conditions: gout. In: RACGP Clinical Guidelines. East Melbourne: RACGP; 2023.
- 14. Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (IL-1 Trap) in patients with cryopyrin-associated periodic syndromes (CAPS). Arthritis Rheum. 2008;58(8):2443–2452.
- 15. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32(8):373–379.