📋 Key Information Summary
- Innate immunity provides the first line of host defence against pathogens through non-specific, germline-encoded mechanisms that require no prior antigen exposure.
- Physical barriers (intact skin, mucosal epithelium, mucociliary clearance) and chemical barriers (low pH, antimicrobial peptides, lysozyme) prevent pathogen entry.
- Pattern-recognition receptors (TLRs, NLRs, RLRs, CLRs) on innate immune cells detect conserved pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).
- Key cellular effectors include neutrophils (first responders), monocytes/macrophages, dendritic cells, natural killer (NK) cells, and innate lymphoid cells (ILCs).
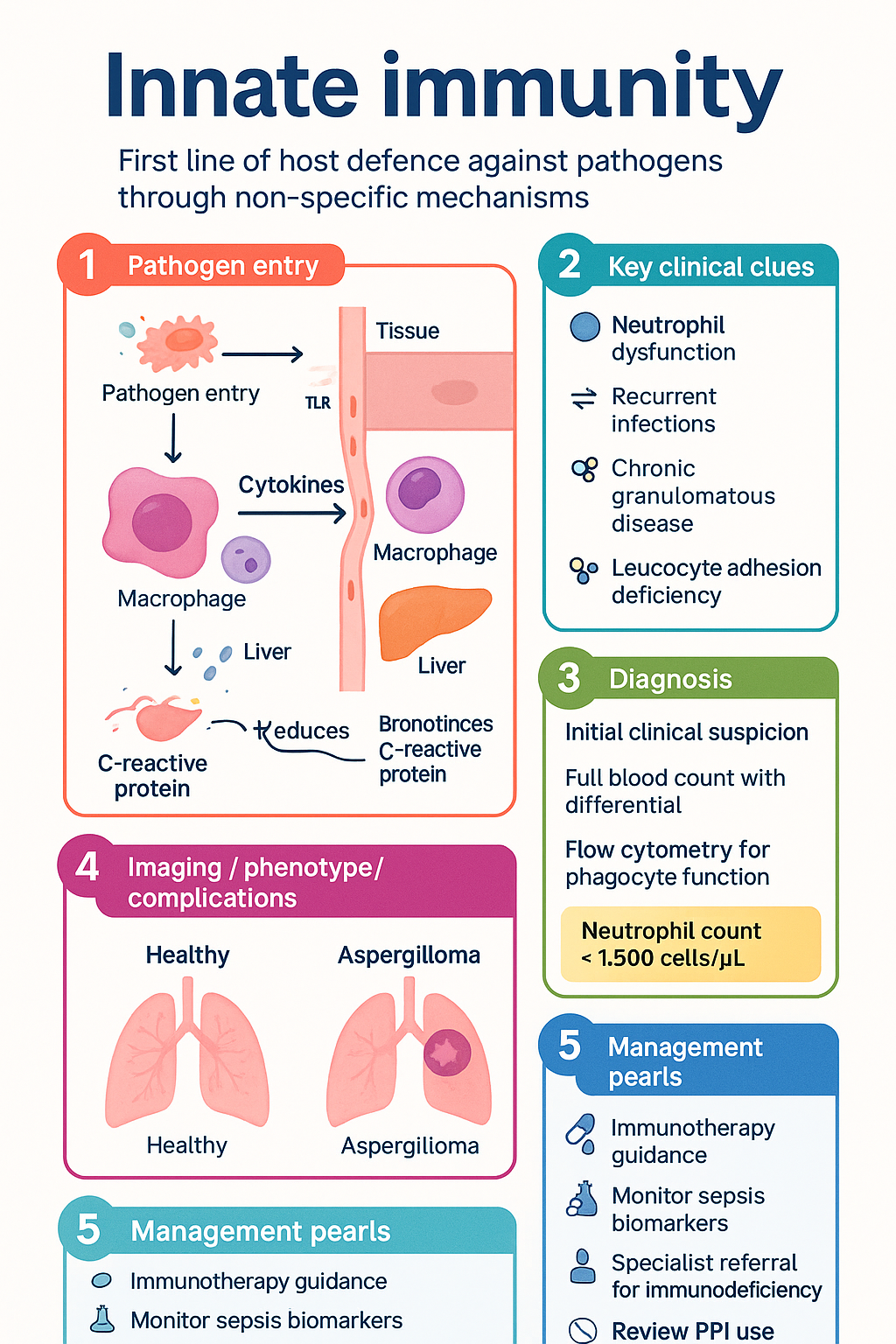
- Neutrophil dysfunction underlies chronic granulomatous disease (CGD), leucocyte adhesion deficiency (LAD), and severe congenital neutropenia — important primary immunodeficiencies in paediatric practice.
- The complement system (>30 proteins) provides opsonisation (C3b), anaphylatoxin generation (C3a, C5a), and membrane attack complex (MAC, C5b-9) formation.
- Complement deficiencies predispose to specific infections: C3 deficiency — severe pyogenic infections; terminal pathway (C5–C9) deficiency — recurrent Neisseria infections.
- Pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and chemokines (CXCL8/IL-8) coordinate the inflammatory response; dysregulation drives sepsis and cytokine storm.
- Aboriginal and Torres Strait Islander populations experience higher rates of infections amenable to innate immune defence (pneumonia, skin infections, rheumatic fever) due to environmental and socioeconomic determinants.
- Investigations include full blood count with differential, CRP, ESR, serum complement levels (CH50, AP50), flow cytometry for phagocyte function (DHR assay), and cytokine panels.
- Understanding innate immunity is essential for interpreting sepsis biomarkers (procalcitonin, presepsin), managing autoinflammatory syndromes (CAPS, FMF, HIDS), and guiding immunotherapy.
Introduction & Australian Context
Innate immunity constitutes the evolutionarily ancient, non-specific arm of the immune system, providing the first line of defence against invading pathogens. Unlike adaptive immunity, innate immune mechanisms are encoded in the germline, require no prior antigen exposure, and respond within minutes to hours of pathogen encounter. These mechanisms encompass physical and chemical barriers, circulating and tissue-resident immune cells, the complement system, and a network of cytokines and mediators that coordinate inflammatory responses.
In Australia, infections related to innate immune dysfunction represent a significant clinical burden. Sepsis accounts for approximately 20,000 hospital admissions annually, with in-hospital mortality rates of 15–20%. Aboriginal and Torres Strait Islander peoples experience disproportionately higher rates of invasive pneumococcal disease, rheumatic fever, and chronic suppurative lung disease, reflecting both innate immune challenges and broader social determinants of health. Primary immunodeficiencies affecting innate pathways — including chronic granulomatous disease, complement deficiencies, and toll-like receptor signalling defects — are increasingly recognised through expanded newborn screening programmes and genetic testing available via Medicare-funded genomic sequencing (MBS item 73425).
This guideline provides a clinically focused overview of innate immune mechanisms relevant to Australian general practice, emergency medicine, and specialist immunology. It covers the recognition of innate immune deficiency, interpretation of relevant investigations, and therapeutic considerations within the Australian healthcare context.
Physical & Chemical Barriers
Physical and chemical barriers represent the outermost layer of innate defence, preventing pathogen colonisation and entry into sterile tissues. These barriers are continuously active and require no immune cell activation.
Physical Barriers
| Barrier | Mechanism | Clinical Relevance |
|---|---|---|
| Stratified squamous epithelium (skin) | Keratinised layer provides physical impermeability; desquamation removes colonising organisms | Burns, eczema, surgical wounds — major portals of infection |
| Mucosal epithelium | Tight junctions, mucus secretion, and continuous cell turnover | Inflammatory bowel disease disrupts GI barrier; H. pylori overcomes gastric mucus |
| Mucociliary escalator | Ciliated epithelium propels mucus-trapped particles toward pharynx | Defective in cystic fibrosis, primary ciliary dyskinesia, smoking |
| Urine flow | Mechanical flushing of urethra | Catheterisation, urinary retention — increased UTI risk |
| Commensal microbiota | Competitive exclusion of pathogens; production of bacteriocins and short-chain fatty acids | Antibiotic disruption predisposes to C. difficile infection |
Chemical Barriers
| Factor | Source | Action |
|---|---|---|
| Hydrochloric acid (pH 1–2) | Gastric parietal cells | Kills ingested organisms; proton pump inhibitors reduce this defence |
| Fatty acids & lysozyme | Sebaceous glands, sweat, tears, saliva | Disrupt bacterial membranes; lysozyme cleaves peptidoglycan |
| Defensins (α and β) | Paneth cells (intestinal), neutrophils, epithelial cells | Form pores in microbial membranes; Paneth cell dysfunction linked to Crohn's disease |
| Cathelicidin (LL-37) | Neutrophils, epithelial cells | Broad antimicrobial, neutralises LPS, modulates inflammation; vitamin D-regulated |
| Secretory IgA | Mucosal plasma cells (transcytosis) | Immune exclusion — agglutinates pathogens, neutralises toxins |
| Lactoferrin | Neutrophil granules, breast milk, secretions | Sequesters iron, bacteriostatic; also direct bactericidal activity |
Pattern-Recognition Receptors (PRRs)
Innate immune cells and epithelial surfaces express germline-encoded PRRs that detect conserved microbial structures (PAMPs) and endogenous danger signals (DAMPs). PRR engagement triggers intracellular signalling cascades leading to cytokine production, inflammasome activation, and adaptive immune priming.
| PRR Family | Location | Key Ligands | Signalling |
|---|---|---|---|
| TLR1–10 (humans) | Cell surface (TLR1,2,4,5,6,10), endosomal (TLR3,7,8,9) | LPS (TLR4), flagellin (TLR5), dsRNA (TLR3), CpG DNA (TLR9) | MyD88, TRIF → NF-κB, IRF3/7 |
| NLRs (NOD1, NOD2, NLRP3) | Cytoplasm | Muramyl dipeptide (NOD2), iE-DAP (NOD1), diverse PAMPs/DAMPs (NLRP3) | NF-κB; NLRP3 inflammasome → caspase-1 → IL-1β, IL-18 |
| RLRs (RIG-I, MDA5) | Cytoplasm | Viral dsRNA, 5′-triphosphate RNA | MAVS → IRF3/7 → type I IFN |
| CLRs (Dectin-1, DC-SIGN, Mincle) | Cell surface | β-glucan (Dectin-1), mannose (DC-SIGN), trehalose dimycolate (Mincle) | Syk/CARD9 → NF-κB; phagocytosis |
| cGAS-STING | Cytoplasm | Cytosolic dsDNA | cGAMP → STING → TBK1 → IRF3 → type I IFN |
Cellular Components
Innate immune cells are rapidly recruited to sites of infection and tissue damage, where they perform phagocytosis, degranulation, antigen presentation, and cytokine secretion. Many also bridge innate and adaptive immunity.
Neutrophils
Neutrophils are the most abundant circulating leucocytes (2.0–7.5 × 10⁹/L in adults) and the first cells recruited to sites of acute infection. They are short-lived (6–12 hours in circulation) but critical for defence against bacteria and fungi.
Monocytes, Macrophages & Dendritic Cells
Monocytes (0.2–1.0 × 10⁹/L) circulate for 1–3 days before migrating into tissues, where they differentiate into macrophages or dendritic cells. Tissue-resident macrophages (Kupffer cells, alveolar macrophages, microglia) provide sentinel surveillance.
Natural Killer (NK) Cells
NK cells (CD56⁺CD3⁻) are innate lymphocytes that provide rapid cytotoxic responses against virus-infected and tumour cells without prior sensitisation. They are controlled by the balance of activating (NKG2D, NKp46) and inhibitory (KIR, NKG2A) receptors — the "missing-self" hypothesis explains their ability to kill MHC-I-downregulated cells.
| NK Subset | Phenotype | Function |
|---|---|---|
| CD56bright CD16dim | ~10% of circulating NK cells | Cytokine production (IFN-γ, TNF-α); immunoregulatory; dominant in lymph nodes |
| CD56dim CD16bright | ~90% of circulating NK cells | High cytotoxicity; ADCC via CD16 (FcγRIIIa); perforin/granzyme-mediated killing |
Innate Lymphoid Cells (ILCs)
ILCs are the innate counterparts of T helper subsets, residing predominantly in mucosal tissues. They lack rearranged antigen receptors and respond rapidly to epithelial-derived cytokines (IL-25, IL-33, TSLP).
| ILC Subset | TH Analogue | Key Cytokines Produced | Function |
|---|---|---|---|
| ILC1 | TH1 | IFN-γ, TNF-α | Intracellular pathogens (viruses, intracellular bacteria) |
| ILC2 | TH2 | IL-4, IL-5, IL-13 | Helminth defence, allergic inflammation, tissue repair |
| ILC3 | TH17 | IL-17, IL-22 | Mucosal barrier integrity, extracellular bacteria, fungi |
Other Innate Immune Cells
- Mast cells: Tissue-resident (skin, mucosa, perivascular); degranulate on IgE crosslinking (FcεRI) or complement (C3a, C5a) — histamine, heparin, proteases, prostaglandins, leukotrienes. Central to allergic and anaphylactic responses.
- Eosinophils: 0.0–0.5 × 10⁹/L; major basic protein (MBP), eosinophil cationic protein (ECP) — helminth killing, allergic tissue damage. Eosinophilia (≥0.5 × 10⁹/L) warrants investigation for parasitic infection, allergy, eosinophilic disorders, and haematological malignancy.
- Basophils: <0.1 × 10⁹/L; circulating counterpart of mast cells; IgE-dependent activation; IL-4 source in early allergic responses.
- NKT cells: Recognise lipid antigens presented by CD1d; rapid IFN-γ and IL-4 production; bridge innate and adaptive immunity.
Primary Immunodeficiencies Affecting Innate Cells
| Condition | Defect | Key Features | Australian Testing |
|---|---|---|---|
| Chronic Granulomatous Disease (CGD) | NOX2 components (CYBB, CYBA, NCF1/2/4) | Catalase-positive organisms (Staph aureus, Aspergillus, Serratia, Nocardia); granuloma formation | DHR flow cytometry (specialist labs — Westmead, RCH Melbourne, QCH Brisbane) |
| Leucocyte Adhesion Deficiency (LAD) I/II/III | CD18 (LAD-I), fucosylation (LAD-II), kindlin-3 (LAD-III) | Delayed umbilical cord separation, omphalitis, periodontitis, leucocytosis without pus formation | Flow cytometry for CD11b/CD18 expression |
| Severe Congenital Neutropenia (SCN) | ELANE, HAX1, G6PC3, others | ANC <0.5 × 10⁹/L from birth; recurrent skin/ear/lung infections from neonatal period | Serial FBC, genetic panel (MBS item 73425), bone marrow aspirate |
| Specific Granule Deficiency | CEBPE transcription factor | Bilobed neutrophils, absent secondary granules, recurrent skin and lung infections | Peripheral blood film, neutrophil granule staining |
Complement System
The complement system comprises over 30 soluble and membrane-bound proteins that form an enzymatic cascade providing rapid antimicrobial defence. Complement activation occurs via three pathways — classical, lectin, and alternative — all converging at C3 convertase formation and leading to opsonisation, inflammation, and direct pathogen lysis.
Activation Pathways
Terminal Pathway & Membrane Attack Complex (MAC)
All three pathways converge to generate C3 convertase, which cleaves C3 into C3a (anaphylatoxin) and C3b (opsonin). C3 convertase combines with C3b to form C5 convertase, cleaving C5 into C5a (potent anaphylatoxin and chemotaxin) and C5b. C5b sequentially recruits C6, C7, C8, and multiple C9 molecules to form the membrane attack complex (MAC, C5b-9), creating transmembrane pores that lyse target cells.
Biological Functions of Complement
| Function | Mediator(s) | Mechanism & Clinical Relevance |
|---|---|---|
| Opsonisation | C3b, iC3b, C4b | Coat pathogens for enhanced phagocytosis via CR1 (C3b), CR3 (iC3b), CR4 (iC3b). Critical for encapsulated organism clearance. |
| Anaphylatoxins | C3a, C4a, C5a | Mast cell degranulation (histamine), smooth muscle contraction, vascular permeability. C5a is the most potent — recruits and activates neutrophils. |
| Chemotaxis | C5a (primary), C3a | Gradient-directed neutrophil and monocyte migration to infection site. |
| Direct lysis | C5b-9 (MAC) | Osmotic lysis of Gram-negative bacteria, enveloped viruses, and abnormal cells. Deficient in recurrent Neisseria infections. |
| Immune complex clearance | C3b, CR1 | C3b binds immune complexes; CR1 on erythrocytes transports to liver/spleen for removal. Deficiency → SLE-like disease. |
| Adaptive immunity bridge | C3d, CR2 (CD21) | C3d-antigen binding to CR2 on B cells lowers activation threshold by 1000–10,000-fold (linked recognition). |
Complement Regulation
Tight regulation prevents complement-mediated host tissue damage. Key regulators include:
- C1 inhibitor (C1-INH): Serine protease inhibitor; blocks classical and lectin pathway activation. Deficiency causes hereditary angioedema (HAE) — C1-INH concentrate (Berinert®) available via special PBS authority for acute attacks.
- Factor H: Cofactor for Factor I-mediated C3b cleavage; displaces Bb from C3b. Mutations/antibodies → atypical haemolytic uraemic syndrome (aHUS) or C3 glomerulopathy.
- Factor I: Serine protease that cleaves C3b and C4b (requires cofactors: Factor H, MCP, C4BP).
- Decay-accelerating factor (DAF/CD55): Accelerates decay of C3/C5 convertases. Deficient in paroxysmal nocturnal haemoglobinuria (PNH) — treated with eculizumab (Soliris®), PBS Authority Required.
- Membrane cofactor protein (MCP/CD46): Cofactor for Factor I; mutations predispose to aHUS.
- CD59 (protectin): Prevents C9 polymerisation and MAC insertion. Deficient in PNH.
Complement Deficiencies & Clinical Syndromes
| Deficiency | Pathway | Clinical Association |
|---|---|---|
| C1q, C2, C4 | Classical | SLE-like autoimmune disease (C1q deficiency → strongest association); immune complex-mediated glomerulonephritis |
| C3 | All pathways converge | Severe recurrent pyogenic infections (S. pneumoniae, H. influenzae, Neisseria); glomerulonephritis; most severe complement deficiency |
| C5, C6, C7, C8, C9 | Terminal (MAC) | Recurrent Neisseria meningitidis and N. gonorrhoeae infections (1,000–10,000-fold increased risk) |
| MBL | Lectin | Increased susceptibility to infections in infancy and immunocompromised; common polymorphism (~5% homozygous deficient) |
| Factor H, Factor I, MCP | Regulatory | Atypical HUS; C3 glomerulopathy; age-related macular degeneration (Factor H polymorphisms) |
Investigations — Complement Assessment
Cytokines & Mediators
Cytokines are small signalling proteins that coordinate the innate immune response, regulate inflammation, and bridge to adaptive immunity. They act in autocrine, paracrine, and endocrine fashions, forming complex networks with synergistic, antagonistic, and pleiotropic effects. Dysregulated cytokine production underlies sepsis, cytokine release syndrome (CRS), autoinflammatory diseases, and chronic inflammatory conditions.
Pro-inflammatory Cytokines
| Cytokine | Primary Sources | Key Functions | Clinical Relevance |
|---|---|---|---|
| TNF-α | Macrophages, monocytes, T cells, mast cells | Master pro-inflammatory cytokine: endothelial activation (E-selectin, ICAM-1), leucocyte recruitment, fever, acute-phase response, apoptosis; systemic effects: hypotension, DIC at high levels | Elevated in sepsis; target of biologics (adalimumab [Humira®, PBS], etanercept [Enbrel®], infliximab [Remicade®]). Anti-TNF therapy increases TB reactivation risk — mandatory screening pre-treatment (MBS item 69343). |
| IL-1β | Macrophages, dendritic cells, epithelial cells | Fever, acute-phase protein induction, neutrophil activation, T-cell co-stimulation. Processed by caspase-1 via NLRP3 inflammasome. | Target in autoinflammatory diseases: anakinra (Kineret®, PBS Authority Required for CAPS/FMF), canakinumab (Ilaris®), rilonacept. IL-1β drives cytokine storm in CRS and severe COVID-19. |
| IL-6 | Macrophages, T cells, fibroblasts, endothelium | Acute-phase response (CRP, fibrinogen, hepcidin synthesis by liver), B-cell differentiation, TH17 differentiation, fever | Major driver of CRS; tocilizumab (Actemra®, PBS Authority Required for CRS) is an anti-IL-6R antibody. IL-6 correlates with disease severity in sepsis and RA. |
| IL-12 | Macrophages, dendritic cells | Drives TH1 differentiation and IFN-γ production by T cells and NK cells; critical for intracellular pathogen defence | IL-12/IL-23 p40 deficiency (Mendelian susceptibility to mycobacterial disease, MSMD) — disseminated BCG/NTM infections |
| IL-18 | Macrophages, epithelial cells | Synergises with IL-12 for IFN-γ production; processed by inflammasome (caspase-1) | Elevated in macrophage activation syndrome (MAS/HLH); IL-18-binding protein (tadekinig alfa) in clinical trials |
| Type I Interferons (IFN-α/β) | Plasmacytoid DCs (IFN-α), most nucleated cells (IFN-β) | Antiviral state: MHC-I upregulation, PKR activation, OAS/RNase L pathway, NK cell activation. Induced via TLR3/7/8/9, RIG-I, cGAS-STING. | Anti-IFN-α autoantibodies cause life-threatening viral infections (COVID-19, influenza). IFN-α (Pegasys®, PBS Authority Required for hepatitis B/C) is therapeutic in viral hepatitis. |
Anti-inflammatory Cytokines
| Cytokine | Function | Therapeutic Relevance |
|---|---|---|
| IL-10 | Potent anti-inflammatory; suppresses macrophage and DC activation, inhibits pro-inflammatory cytokine production, promotes Treg differentiation | IL-10 deficiency → very-early-onset IBD (VEO-IBD); IL-10R mutations require haematopoietic stem cell transplant |
| TGF-β | Immunosuppression, wound healing, fibrosis, Treg induction, IgA class switching | Excessive TGF-β → fibrotic disease (IPF, liver cirrhosis); pirfenidone (Esbriet®, PBS for IPF) partially targets this pathway |
| IL-1Ra | Natural competitive antagonist of IL-1 receptor; limits IL-1β signalling | Recombinant IL-1Ra (anakinra) used therapeutically in autoinflammatory diseases, gout flares, and Still's disease |
| IL-35 | Produced by Tregs; suppresses T-cell proliferation and TH17 differentiation | Emerging target in autoimmune and inflammatory disease management |
Chemokines
Chemokines are small chemoattractant cytokines that direct leucocyte migration. They are classified by cysteine residue arrangement (CXC, CC, CX3C, C).
| Chemokine | Receptor | Target Cell | Role |
|---|---|---|---|
| CXCL8 (IL-8) | CXCR1, CXCR2 | Neutrophils | Major neutrophil chemoattractant; rapid neutrophil recruitment to sites of bacterial infection |
| CCL2 (MCP-1) | CCR2 | Monocytes, memory T cells | Monocyte recruitment to inflamed tissues; implicated in atherosclerosis, obesity-related inflammation |
| CCL3 (MIP-1α), CCL5 (RANTES) | CCR1, CCR5 | Monocytes, T cells, NK cells | Inflammatory cell recruitment; CCR5 is an HIV co-receptor — maraviroc (Celsentri®, PBS) blocks this |
| CCL19, CCL21 | CCR7 | Mature DCs, naïve T cells | Direct DC and T-cell homing to lymph nodes for adaptive immune priming |
| CXCL12 (SDF-1) | CXCR4 | Haematopoietic stem cells, lymphocytes | Stem cell niche retention; plerixaabor (Mozobil®, PBS Authority Required) mobilises HSCs for transplant |
Other Key Mediators of Innate Immunity
- Prostaglandins & Leukotrienes: Arachidonic acid metabolites (via COX-1/2 and 5-LOX pathways). PGE₂ causes vasodilation, fever, pain sensitisation. LTB₄ is a potent neutrophil chemoattractant. Cysteinyl leukotrienes (LTC₄, LTD₄, LTE₄) cause bronchoconstriction and mucus secretion — target of montelukast (Singulair®, PBS General Benefit).
- Histamine: Released from mast cells and basophils; H1 receptor activation causes vasodilation, increased permeability, pruritus, bronchoconstriction. H1 antihistamines (cetirizine, fexofenadine — PBS General Benefit) are first-line for allergic symptoms.
- Nitric Oxide (NO): Produced by inducible NOS (iNOS/NOS2) in macrophages; antimicrobial via reactive nitrogen species; also vasodilator contributing to septic shock.
- Reactive Oxygen Species (ROS): Superoxide (O₂⁻), hydrogen peroxide (H₂O₂), hypochlorous acid (HOCl) — generated by NADPH oxidase in phagocytes; microbicidal but cause tissue damage in chronic granulomatous inflammation.
- DAMPs (alarmins): HMGB1, ATP, uric acid crystals, heat shock proteins, mitochondrial DNA, IL-33, IL-1α — released from damaged/necrotic cells; activate PRRs and sterile inflammation. Relevant to trauma, ischaemia-reperfusion injury, and gout (monosodium urate crystals activate NLRP3 inflammasome).
Cytokine Storm & Clinical Syndromes
- Sepsis — balanced vs. hyperinflammatory phenotypes
- Haemophagocytic lymphohistiocytosis (HLH) — primary (genetic) or secondary (infection, malignancy, autoimmune)
- Cytokine release syndrome (CRS) — CAR-T cell therapy, bispecific antibodies (tocilizumab PBS-listed for CRS management)
- Severe COVID-19 — tocilizumab and baricitinib demonstrated mortality benefit in RECOVERY and ACTT-2 trials
- Macrophage activation syndrome (MAS) — complication of systemic JIA and SLE
Clinical Approach to Suspected Innate Immune Deficiency
Recognition of innate immune deficiency in primary care depends on pattern recognition of recurrent, severe, or unusual infections. The Australasian Society of Clinical Immunology and Allergy (ASCIA) provides referral guidelines.
Red Flags for Referral to Immunology
- ≥2 systemic bacterial infections in 12 months (septicaemia, deep abscesses, osteomyelitis, pneumonia requiring hospitalisation)
- Infections with unusual organisms (Aspergillus, Nocardia, Serratia, atypical mycobacteria, Pneumocystis jirovecii) in an immunocompetent-appearing host
- Failure to thrive with recurrent infections in infants
- Recurrent meningococcal disease (consider terminal complement deficiency)
- Family history of primary immunodeficiency or consanguinity
- Persistent oral candidiasis or dermatophytosis beyond childhood (consider STAT1 GOF, CARD9 deficiency)
- Severe viral infections (disseminated HSV/VZV, fatal EBV) — consider inborn errors of type I IFN immunity
Primary Care Investigations (Before Specialist Referral)
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Murphy K, Weaver C. Janeway's Immunobiology. 10th ed. New York: Garland Science; 2022.
- 2. Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 10th ed. Philadelphia: Elsevier; 2022.
- 3. Australasian Society of Clinical Immunology and Allergy (ASCIA). ASCIA Guide to Investigation and Management of Primary Immunodeficiency. Sydney: ASCIA; 2024. Available from: https://www.allergy.org.au
- 4. Tangye SG, Bucciol G, Meyts I. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2024;44(1):22–68.
- 5. Mastaglia FL. Innate immunity and the role of pattern recognition receptors. Aust Prescr. 2021;44(4):118–122.
- 6. Australian Commission on Safety and Quality in Health Care (ACSQHC). Sepsis Clinical Care Standard. Sydney: ACSQHC; 2023. Available from: https://www.safetyandquality.gov.au
- 7. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I — molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262.
- 8. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023 Summary Report. Canberra: AIHW; 2023.
- 9. RHDAustralia (RHD Australia, ARF/RHD writing group). The 2020 Australian guideline for prevention, diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd ed. Darwin: Menzies School of Health Research; 2020.
- 10. Dinn RL, Bhojwani R, Engel M, et al. Inborn errors of immunity in Australia: a national approach to newborn screening. J Clin Immunol. 2023;43(6):1068–1078.
- 11. Bowen AC, Mahé A, Hay RJ, et al. The global epidemiology of impetigo: a systematic review of the population prevalence of impetigo and pyoderma. PLoS One. 2015;10(8):e0136789.
- 12. The RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10285):1637–1645.
- 13. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255–2273.
- 14. Pharmaceutical Benefits Scheme (PBS). Australian Government Department of Health and Aged Care. Available from: https://www.pbs.gov.au. Accessed 2024.