Cholesteatoma is an accumulation of squamous epithelium and keratin debris that usually involves the middle ear and mastoid.
Although benign, it may enlarge and invade adjacent bone.
Often presents with a malodorous ear discharge with associated hearing loss.

Diagnosis is clinical based on history and otoscopic findings. CT scan of the temporal bone, provides lesion definition and extent.
Treatment is surgical removal. Adjunctive topical antimicrobial treatment may help reduce acute symptoms preoperatively.
Complications include hearing loss, recurrence, meningitis, facial palsy, and a labyrinthine fistula.
Definition
Cholesteatoma is defined as the presence of keratinising squamous epithelium within the middle ear, or in other pneumatised areas of the temporal bone.
This keratinising epithelium exhibits independent growth, leading to expansion and to resorption of underlying bone.
Focal erosion of external canal bone with accumulation of keratin is called external canal cholesteatoma.
Epidemiology
The annual incidence of cholesteatoma ranges from 9 to 12.6 cases per 100,000 in adults and from 3 to 15 cases per 100,000 in children.
This varies both worldwide and with the population studied. In Scotland, an annual incidence of 13 per 100,000 of the population was noted for the period 1966 to 1986.
In Iowa, US, the overall incidence is 7 per 100,000 per year in children.[11] Congenital cholesteatoma is less common. For example, a study from Denmark reports an annual incidence of surgical treatment for congenital cholesteatoma of 0.12 per 100,000, compared to 2.3 per 100,000 for acquired cholesteatoma.
Incidence may be affected by medical practice. The use of grommets or aural ventilation tubes in patients with chronic otitis media was associated with a fall in the incidence of cholesteatoma surgery in Finland and Israel.
However, no change was noted in the incidence of disease in Scotland and Ireland, with these treatments.
There are fewer data on prevalence, but in Jerusalem it has been quoted as high as 70 per 100,000 children.[16] In Australian Aboriginal children, a prevalence of 50 per 100,000 in a study of 7362 ears has been reported.
Both males and females are affected, with a ratio of 3:2. Cholesteatoma in children has been found to affect the eustachian tube, anterior mesotympanum, retrolabyrinthine cells, and mastoid tip more than in adults.
Clinical and histological evidence suggest that cholesteatoma in children tends to be more aggressive.
Aetiology
Cholesteatoma may be considered acquired or congenital.
Acquired cholesteatoma occurs in several ways. In many cases, it is due to retraction of an area of the pars flaccida with or without associated atrophy of the pars tensa.
The epithelium becomes trapped and infected, and proliferates to form a cholesteatoma.
Alternatively, squamous epithelium may migrate through a defect in the tympanic membrane, or the cholesteatoma may form due to implantation of viable keratinocytes into the middle ear cleft following otological surgery or after a traumatic blast injury.
Children with a cleft palate, craniofacial abnormality or a chromosomal disorder (e.g., Turner's or Down's syndrome), have an increased risk of developing cholesteatoma.
This increased risk is secondary to poor eustachian tube function.
Congenital cholesteatoma is considered to be present if there is no history of previous ear surgery and no perforation or retraction of the tympanic membrane.
It is believed to arise from developmental epidermoid rests present before birth that persist in the middle ear space.
Alternative theories include: invagination of squamous epithelium from the developing ear canal; seeding of the middle ear by squamous cells in the amniotic fluid; epithelial ingrowth from the surface of the tympanic membrane after infection; and micro-perforation.
Staging of cholesteatoma
The European Academy of Otology and Neurotology (EAONO) and Japanese Otological Society (JOS) working group developed a staging system that applies to four categories of middle ear cholesteatoma: pars flaccida cholesteatoma; pars tensa cholesteatoma; congenital cholesteatoma; and cholesteatoma secondary to a pars tensa perforation.
This staging system does not apply to petrous bone cholesteatoma.
This system may be used for evaluating initial pathology in a standardised fashion. It may also be used to adjust for the severity of the condition during outcome evaluations, as well as providing information that is useful for counselling patients.
Their findings are outlined below:
Stage 1: Localised cholesteatoma (described based on the primary site of origin)
- The attic for pars flaccida cholesteatoma
- The tympanic cavity for pars tensa cholesteatoma
- Congenital cholesteatoma
- Cholesteatoma secondary to a pars tensa perforation.
Stage 2: Cholesteatoma involving two or more sites.
Stage 3: Cholesteatoma with extracranial complications or pathological conditions.
These include: facial palsy; labyrinthine fistula; labyrinthitis; postauricular abscess or fistula; zygomatic abscess; neck abscess; canal wall destruction more than half the length of the bony ear canal; destruction of the tegmen, with a defect that requires surgical repair; and adhesive otitis.
Stage 4: Cholesteatoma with intracranial complications.
These include: purulent meningitis, epidural abscess, subdural abscess, brain abscess, sinus thrombosis, and brain herniation into the mastoid cavity.
↚
Pathophysiology
A retraction pocket is an area of invagination of the tympanic membrane that becomes pulled into the middle ear space because of the negative pressure (vacuum-like) effect of eustachian tube dysfunction.
These pockets are initially self-cleansing.
They are graded or staged according to the degree of severity:
- Stage 1: retracted membrane
- Stage 2: retraction onto the incus
- Stage 3: middle ear atelectasis
- Stage 4: adhesive otitis media.
They are often found as the initial step in acquired cholesteatoma in adults or children; it is difficult to predict which pockets will develop into a cholesteatoma.
Clinical and histological evidence suggest that cholesteatoma in children tends to be more aggressive.
The neck of the pocket may narrow and trap squamous cells with keratin accumulation and retention; these proliferate, leading to expansion and cholesteatoma formation.
This may occur as papillae migrating through a temporary defect in the pars flaccida or around a pars tensa perforation (marginal perforation).
Less commonly there is proliferation of the basal layers of keratinising epithelium of the pars flaccida.
Bacterial infection and super-infection of the trapped debris form a biofilm and cause chronic infection and epithelial proliferation, leading to further lesion expansion and cholesteatoma formation. There is often infection with Pseudomonas aeruginosa species.
Once formed, the cholesteatoma sac is associated with enzymatic bony destruction due to cytokine-induced inflammatory changes, with activation of osteoclasts and lysozymes.
Cholesteatoma is often associated with destruction of the ossicles causing a conductive hearing loss, and may be associated with destruction of the semicircular canals (with resulting vertigo), cochlea (with resulting sensorineural hearing loss), and facial canal (with resulting facial palsy).
Congenital cholesteatoma may present as an epidermoid cyst behind an intact tympanic membrane. It is often found in the anterior superior aspect of the middle ear.
This frequently arises above the eustachian tube orifice and obstructs it early in its course, leading to a middle ear effusion.
With time, however, the cyst can perforate, presenting as an acquired cholesteatoma.
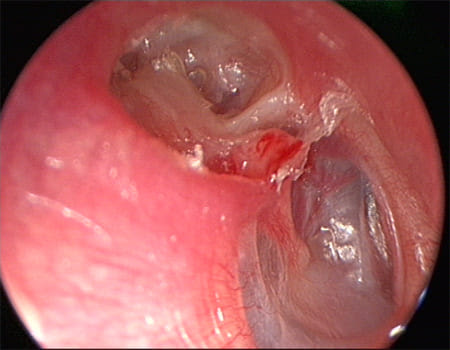 |
| (upper part of the middle ear) cholesteatoma - Retraction pocket in attic |

Aetiopathological classification
- Primary: occurs as a consequence of retraction pocket formation within the tympanic membrane subsequent to eustachian tube dysfunction, with invagination of squamous cells into the middle ear.
- Secondary: occurs due to migration of squamous epithelium through an established tympanic membrane defect (marginal perforation), or due to implantation of viable keratinocytes into the middle ear cleft following otological surgery or after a traumatic blast injury.
Case history
Diagnostic approach
History and examination
Audiology
Imaging


Microbiology
Ear cultures are obtained mainly in patients who present with an aural discharge unresponsive to antimicrobial therapies. Swabs from the ear demonstrate bacterial infection, commonly Pseudomonas aeruginosa, and may demonstrate other pathogens such as Staphylococcus aureus and anaerobic bacteria.
↚
Management approach
Patients suspected of having cholesteatoma should be referred to an ear, nose, and throat surgeon on a semi-urgent basis.
The American Academy of Pediatrics recommends that infants and children should be referred to a paediatric otolaryngologist.
If facial palsy is present with suspected cholesteatoma, those patients should be referred urgently because early treatment is associated with better outcomes and treatment delays can result in a poorer prognosis.
The definitive treatment of cholesteatoma is surgery.
The main goal is to remove the disease; provide a dry, safe ear that does not discharge; and prevent potential complications. Treatment is also aimed at improving the hearing threshold.
This may not always be possible at initial surgery but may be possible following a second procedure. There are different surgical approaches: the canal wall up mastoidectomy or the canal wall down mastoidectomy. The use of endoscopy has been shown to positively impact the management of cholesteatoma and its use is recommended during surgery.
Utilisation of high definition visualisation, such as a 4K magnification endoscope and narrow band imaging filter has been shown to improve visualisation of tissue based on varying degrees of vascularity allowing for better differentiation between pathology and normal anatomy and this may reduce disease recurrence after surgery.
Controlled hypotension is often required in middle ear surgery.
A study has demonstrated that both magnesium sulfate and remifentanil in combination with sevoflurane can be used for this purpose but that magnesium sulfate gives better postoperative analgesia and reduces shivering and postoperative nausea and vomiting.
Studies have also demonstrated that 8 mg dexamethasone is more effective than 4 mg dexamethasone at controlling postoperative nausea and vomiting and providing analgesia in adult patients after middle ear surgery.
Additionally, the combination of dexamethasone and midazolam may also reduce postoperative nausea and vomiting.
Pre-surgical care
On initial presentation, prior to definitive surgical treatment, it may be appropriate to treat the aural discharge with topical antibiotics.
Agents containing quinolone (e.g., ciprofloxacin and ofloxacin) are effective, in adults and in children, either alone or in combination with a topical corticosteroid.
Aural cleaning may also reduce symptomatic discharge. The ear canal may need to be cleaned of any debris or wax prior to the use of topical ear drops.
Patients who have severe swelling of the ear canal may have difficulty applying ear drops. A wick should be inserted in the ear canal to allow for drug delivery. In some patients, debridement of granulation tissue may be necessary.
Canal wall up mastoidectomy (combined approach tympanoplasty)
This procedure allows removal of cholesteatoma but leaves the canal wall intact.
The procedure involves removal of the mastoid air cells lateral to the facial nerve and otic capsule, leaving the posterior and superior parts of the external canal wall intact.
It is often preferred for children, as it avoids long-term complications of a mastoid cavity. However, it necessitates a second-look procedure after 9 to 12 months to examine for residual/recurrent disease. A meta-analysis found an increased incidence of postoperative cholesteatoma when using an intact wall approach rather than canal wall down approach.
Alternatively, a non-echo-planar diffusion-weighted MRI may be used in some patients.
There continues to be debate regarding which type of MRI is best to examine for recurrent cholesteatoma.
Some authors advocate routine MRI scans (such as non-echo-planar, fast-spin echo, or diffusion weighted sequences) for follow-up, but caution that a negative scan may not be completely accurate as residual or recurrent disease may not yet be detectable.
Non-echo-planar imaging was found to be more reliable compared with echo-planar imaging in identifying residual/recurrent cholesteatoma in one systematic review.
Another systematic review found that non-echo-planar diffusion-weighted MRI is highly sensitive and specific in identifying middle-ear cholesteatoma.
Patients will require continued follow-up.
Canal wall down mastoidectomy
This procedure aims to remove the disease by drilling from the attic wall posteriorly. The size of the resultant cavity will depend on the extent of the cholesteatoma.
A less invasive procedure resulting in a minimal cavity is called an atticotomy or atticoantrotomy; a more invasive procedure resulting in a larger cavity is called a modified radical mastoidectomy.
A canal wall down procedure may allow direct examination of the cavity for recurrence, but if the attic has been reconstructed a second-look procedure or a non-echo-planar diffusion-weighted MRI may be necessary to examine the middle ear for recurrent disease.