📋 Key Information Summary
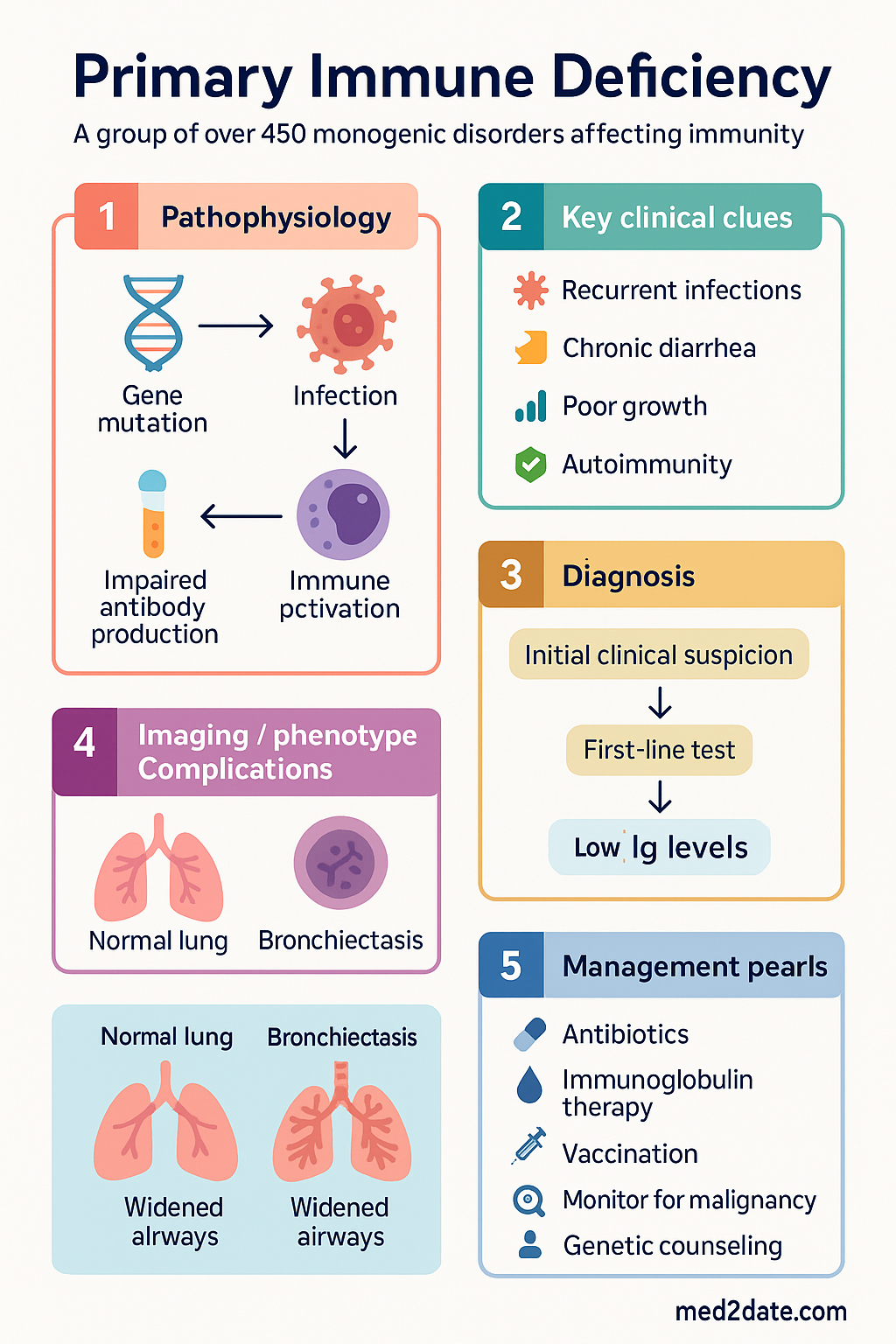
- Primary immunodeficiencies (PIDs) are a group of over 450 monogenic disorders affecting innate or adaptive immunity, leading to recurrent, severe, or unusual infections.
- Suspect PID in patients with ≥2 significant bacterial infections in 1 year, recurrent unusual infections, or family history of PID.
- Classification is based on the primary immune component affected: antibody deficiencies, combined T- and B-cell deficiencies, phagocytic defects, complement defects, and other well-defined syndromes.
- Antibody deficiencies (e.g., CVID, XLA) are the most common symptomatic PIDs, presenting with recurrent sinopulmonary infections.
- T-cell deficiencies (e.g., DiGeorge syndrome) present in infancy with viral, fungal, and opportunistic infections and failure to thrive.
- Severe Combined Immunodeficiency (SCID) is a paediatric emergency requiring urgent referral to a specialist immunology centre.
- First-line investigations include serum immunoglobulins (IgG, IgA, IgM), specific antibody responses, lymphocyte subsets, and CH50.
- Immunoglobulin replacement therapy (intravenous or subcutaneous) is the cornerstone of treatment for most antibody deficiencies.
- Live vaccines (e.g., BCG, OPV, MMR) are contraindicated in suspected or confirmed T-cell or combined deficiencies.
- Refer to a clinical immunologist for diagnostic confirmation and management; early diagnosis improves outcomes.
- Aboriginal and Torres Strait Islander peoples have a higher burden of infectious disease; maintain a high index of suspicion for PID.
- Monitor for non-infectious complications, including autoimmunity, granulomatous disease, and lymphoproliferative disorders.
Introduction & Australian Epidemiology
Primary immunodeficiencies (PIDs) are a heterogeneous group of inherited disorders of immune function, broadly classified by the component of the immune system that is defective. They result in impaired host defence, leading to increased susceptibility to infections, autoimmunity, inflammation, and malignancy. While individually rare, their combined prevalence is estimated at 1 in 1,200 to 1 in 2,000 individuals in Australia.
In Australia, the Australasian Society of Clinical Immunology and Allergy (ASCIA) and the Australian National PIDs Registry have been pivotal in improving diagnosis and management. Antibody deficiencies account for over 50% of diagnosed cases. There is significant under-diagnosis, particularly in adult-onset conditions like Common Variable Immunodeficiency (CVID).
Classification
The International Union of Immunological Societies (IUS) classifies PIDs into ten major categories. The most clinically relevant groups for general practice are:
| Major Category | Examples | Key Defect |
|---|---|---|
| Combined T- and B-cell immunodeficiencies | Severe Combined Immunodeficiency (SCID), Ataxia Telangiectasia | T-cell development/function |
| Predominantly antibody deficiencies | X-linked Agammaglobulinaemia (XLA), Common Variable Immunodeficiency (CVID) | Antibody production |
| Diseases of immune dysregulation | IPEX syndrome | Regulatory T-cell function |
| Congenital defects of phagocyte number or function | Chronic Granulomatous Disease (CGD) | Microbial killing |
| Complement deficiencies | C2 deficiency | Opsonisation/MAC |
Antibody Deficiencies
This is the most common symptomatic group of PIDs. Patients present with recurrent bacterial infections, particularly of the respiratory tract.
Common Variable Immunodeficiency (CVID)
A heterogeneous disorder with onset typically in the 2nd-4th decade. Marked by reduced serum IgG and IgA and/or IgM, with poor vaccine responses.
X-linked Agammaglobulinaemia (XLA, Bruton's)
An X-linked recessive disorder due to BTK mutations, resulting in absent mature B-cells and profoundly low immunoglobulins. Presents in male infants after 6 months of age with recurrent sinopulmonary and enteroviral infections. Requires lifelong immunoglobulin replacement.
T Cell Deficiencies
T-cell defects impair cellular immunity, leading to susceptibility to viral, fungal, protozoal, and intracellular bacterial infections. They are often severe and present early in life.
DiGeorge Syndrome (22q11.2 Deletion)
The most common T-cell deficiency. Characterised by thymic hypoplasia/aplasia, congenital heart defects, hypoparathyroidism, and dysmorphic facies. Immunological severity varies from complete DiGeorge (no T-cells, SCID-like) to partial DiGeorge.
Other T-cell Deficiencies
- CHARGE Syndrome: Includes coloboma, heart defects, choanal atresia, growth retardation, genital abnormalities, and ear anomalies. Variable immune deficiency.
- CARTilage-Hair Hypoplasia: Short-limbed dwarfism with combined T- and B-cell deficiency in some.
Combined Deficiencies
These disorders affect both humoral (B-cell) and cell-mediated (T-cell) immunity, resulting in the most severe phenotypes.
Severe Combined Immunodeficiency (SCID)
A paediatric emergency. Infants typically present by 3-6 months with failure to thrive, persistent diarrhoea, oral thrush, and severe, recurrent, or opportunistic infections (e.g., Pneumocystis jirovecii pneumonia). Newborn screening using the T-cell Receptor Excision Circle (TREC) assay is now performed in all Australian states.
Wiskott-Aldrich Syndrome
X-linked disorder with eczema, thrombocytopenia (small platelets), and combined immunodeficiency. Presents with bleeding, recurrent infections, and increased risk of autoimmunity and lymphoma.
Investigations
Initial assessment can be performed in primary care, but interpretation and definitive testing require clinical immunology input.
Management Principles
Management is guided by the specific PID diagnosis and involves preventing and treating infections, replacing immune function, and managing complications.
Infection Prevention
- Administer all scheduled inactivated vaccines on time. Live vaccines are contraindicated in most PIDs.
- Consider prophylactic antibiotics (e.g., cotrimoxazole 480 mg PO daily or 3 times/week) in patients with recurrent infections or specific defects like CGD.
- Prompt and aggressive treatment of breakthrough infections.
Immunoglobulin Replacement
As detailed in the Antibody Deficiencies section, this is the mainstay for humoral deficiencies. The goal is to maintain a trough IgG level > 5-7 g/L (target varies by patient and infection history).
Definitive Therapies
- Haematopoietic Stem Cell Transplant (HSCT): Curative for SCID, Wiskott-Aldrich, and other severe combined defects. Performed at specialised paediatric centres (e.g., Sydney Children's Hospital, Royal Children's Hospital Melbourne).
- Gene Therapy: Available for specific PIDs (e.g., ADA-SCID) as part of clinical trials or approved therapies at designated centres.
Special Populations
🇦🇺 Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Australasian Society of Clinical Immunology and Allergy (ASCIA). ASCIA Guide to the Investigation of Primary Immunodeficiency. 2023.
- 2. Tangye SG, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022.
- 3. Australian Institute of Health and Welfare (AIHW). Immunisation and vaccine preventable disease. 2023.
- 4. National Blood Authority. National Policy: Access to Immunoglobulin in Australia. 2022.
- 5. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007.
- 6. Slatter MA, Gennery AR. Haematopoietic cell transplantation for primary immunodeficiencies. Br J Haematol. 2021.
- 7. Kwan A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014.
- 8. Department of Health (Australian Government). The Australian Immunisation Handbook. 2023.
- 9. Australian Government Department of Health. Life Saving Drugs Program (LSDP). 2024.
- 10. Bousfiha A, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022.