📋 Key Information Summary
- SCID is a paediatric emergency — infants present within the first 3–6 months of life with recurrent, severe, or opportunistic infections and progressive lymphopenia.
- Without definitive treatment (haematopoietic stem cell transplantation or gene therapy), SCID is uniformly fatal, usually by 12–24 months of age.
- The most common forms are X-linked SCID (IL2RG mutation, ~50% of cases) and adenosine deaminase (ADA) deficiency (~15%).
- X-linked SCID presents with absent T cells, present but non-functional B cells, and absent or low NK cells.
- ADA-SCID presents with absent T, B, and NK cells (T−B−NK− phenotype) plus systemic toxic metabolite effects on non-immune organs.
- Newborn screening using the T-cell receptor excision circle (TREC) assay is now routine in all Australian states and territories, enabling presymptomatic diagnosis.
- Absolute lymphocyte count (ALC) < 3 × 10⁹/L in infancy is a simple, cost-effective screening hint — always request flow cytometry (CD3, CD4, CD8, CD19, CD16/56) if ALC is low.
- All live vaccines (including BCG, rotavirus, OPV, MMR, varicella) are absolutely contraindicated in suspected or confirmed SCID.
- Early haematopoietic stem cell transplant (HSCT) before 3.5 months of age and before any infection achieves > 90% survival.
- Autologous gene therapy using lentiviral vectors is now TGA-approved for ADA-SCID and is an important option when no matched donor is available.
- Reverse isolation, antimicrobial prophylaxis (co-trimoxazole, fluconazole, antivirals), and IVIg replacement are essential bridging measures pre-transplant.
- Aboriginal and Torres Strait Islander infants in remote areas face diagnostic delays — enhanced community awareness and PIP QI targets support earlier referral.
Introduction & Australian Epidemiology
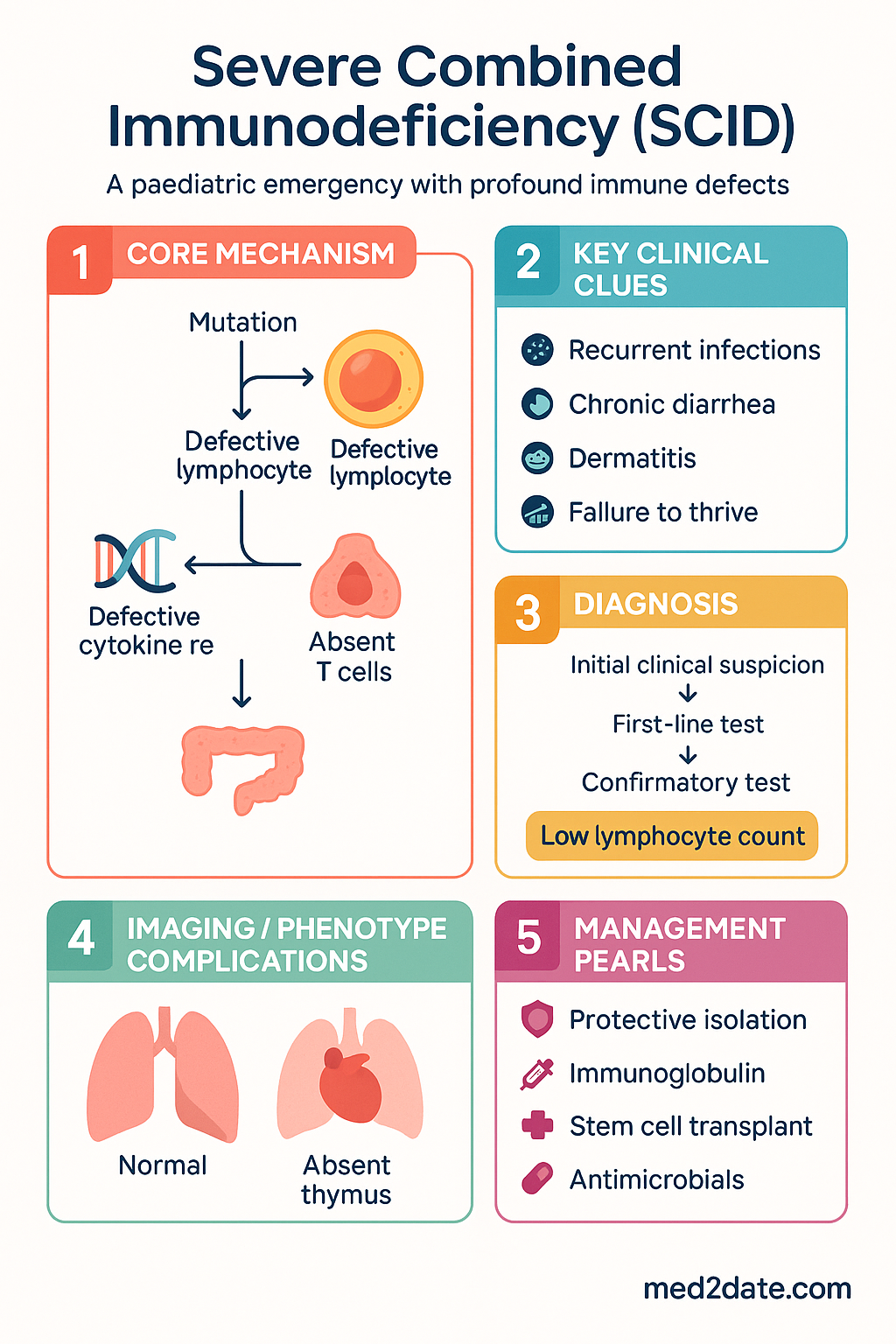
Severe Combined Immunodeficiency (SCID) represents the most severe form of primary immunodeficiency, characterised by profound defects in both cellular and humoral immunity. It is a paediatric emergency: without definitive immune reconstitution, affected infants typically succumb to overwhelming infection within the first two years of life.
SCID encompasses a heterogeneous group of monogenic disorders, all of which disrupt T-cell development and, variably, B-cell and NK-cell maturation. Over 20 causative genes have been identified, with X-linked SCID (IL2RG mutations) and adenosine deaminase deficiency (ADA mutations) together accounting for approximately 65% of cases worldwide.
Australian incidence: SCID affects approximately 1 in 58 000–75 000 live births, translating to an estimated 4–6 new cases per year nationally. Since the introduction of newborn screening across all Australian jurisdictions (completed by 2023), case ascertainment has improved, with presymptomatic diagnosis now the expected standard.
Key Australian referral centres include the Royal Children's Hospital Melbourne, the Children's Hospital at Westmead Sydney, Queensland Children's Hospital Brisbane, and Perth Children's Hospital. Telehealth multidisciplinary teams (MDTs) enable early specialist involvement for patients in regional and remote areas.
Types of SCID
SCID is classified by the immunological phenotype (T, B, NK cell presence) and the underlying genetic defect. The two types discussed in detail below represent the most common subtypes encountered in Australian paediatric practice.
| Feature | X-linked SCID (IL2RG) | ADA-SCID |
|---|---|---|
| Gene / locus | IL2RG — Xq13.1 (encodes common γ chain, γc) | ADA — 20q13.12 (adenosine deaminase) |
| Frequency | ~50% of all SCID; exclusively males | ~15% of all SCID; autosomal recessive |
| Immunophenotype | T− B+ NK− (most common) or T− B+ NK+ | T− B− NK− |
| Mechanism | Loss of cytokine signalling via IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 receptors → absent T/NK cell development; B cells present but non-functional without T-cell help | Accumulation of deoxyadenosine and dATP → toxic to lymphocytes; also causes hepatotoxicity, skeletal abnormalities, neurodevelopmental delay |
| Extra-immune features | Generally none | Costochondral flaring, rachitic changes, hepatomegaly, auditory neurotoxicity, neurodevelopmental delay |
| Maternal engraftment | Common; maternal T cells may cause GVHD-like rash | Less common |
| Inheritance | X-linked recessive (carrier testing for mothers/sisters) | Autosomal recessive (25% recurrence risk per pregnancy) |
Other SCID Genotypes
Less common but clinically important forms include:
- JAK3 deficiency — autosomal recessive; T−B−NK− phenotype (mimics X-linked SCID but affects both sexes)
- RAG1/RAG2 deficiency — autosomal recessive; T−B−NK+; Omenn syndrome variant presents with erythroderma, eosinophilia, lymphadenopathy, hepatosplenomegaly
- Artemis deficiency (DCLRE1C) — radiosensitive SCID; important for pre-transplant conditioning (avoid irradiation-based regimens)
- IL7Rα deficiency — autosomal recessive; T−B+NK+ phenotype
- CD3δ, CD3ε, CD3ζ deficiencies — rare; T−B+NK+ phenotype
- PNP deficiency — purine nucleoside phosphorylase; neurological features prominent
Clinical Features
Symptoms typically begin by 3–6 months of age, coinciding with waning maternal antibody. The clinical presentation is characterised by recurrent, severe, persistent, or opportunistic infections in a previously well-appearing infant.
Classic Presenting Features
- Recurrent sinopulmonary infections — > 2 episodes of pneumonia, otitis media, or sinusitis in 6 months
- Chronic diarrhoea — often persistent and watery; may be due to opportunistic organisms (Cryptosporidium, rotavirus, norovirus)
- Failure to thrive — poor weight gain, crossing centiles downward on WHO growth charts
- Oral/perianal thrush — persistent, extending beyond the oral mucosa, refractory to standard antifungal therapy
- Opportunistic infections — PJP pneumonia, disseminated BCG-osis (in vaccinated infants), invasive candidiasis, CMV viraemia
- Skin manifestations — generalised rash (GVHD from maternal engraftment or Omenn syndrome), eczematous dermatitis
- Hepatosplenomegaly — particularly in Omenn syndrome or ADA-SCID
• Any infant with disseminated BCG-osis after BCG vaccination
• Persistent candidiasis in an infant < 6 months
• Lymphopenia (ALC < 3 × 10⁹/L) with any infection
• Positive newborn screen for low TRECs
• Family history of SCID or known carrier status
ADA-SCID–Specific Features
In addition to infectious manifestations, ADA deficiency causes toxicity from accumulated deoxyadenosine and deoxyadenosine triphosphate (dATP) in non-immune tissues:
- Skeletal abnormalities — costochondral junction widening, cupped ribs, rachitic changes visible on chest X-ray
- Hepatic dysfunction — transaminitis, hepatomegaly
- Neurodevelopmental delay — sensorineural hearing loss, motor delay, cognitive impairment
- Auditory toxicity — high-frequency hearing loss; audiometry recommended at diagnosis
Omenn Syndrome
A phenotypic variant seen in hypomorphic RAG1/2, Artemis, or IL7Rα mutations. Features include generalised erythroderma, alopecia, lymphadenopathy, hepatosplenomegaly, eosinophilia, and elevated IgE. Maternal T-cell engraftment can produce a similar picture in classical SCID.
Diagnosis & Newborn Screening
Newborn Screening (NBS)
All Australian states and territories now include the T-cell receptor excision circle (TREC) assay as part of the newborn bloodspot screening programme. TRECs are a biomarker of thymic output; low or absent TRECs indicate reduced naive T-cell production.
| Jurisdiction | NBS Programme | Status |
|---|---|---|
| New South Wales / ACT | NSW Newborn Screening Programme | TREC included |
| Victoria | Victorian Clinical Genetics Services (VCGS) | TREC included |
| Queensland | Queensland Health NBS | TREC included |
| South Australia | SA Pathology | TREC included |
| Western Australia | PathWest | TREC included |
| Tasmania | Royal Hobart Hospital NBS | TREC included |
| Northern Territory | NT Pathology | TREC included |
An abnormal TREC result triggers immediate recall for confirmatory testing. In addition to SCID, low TRECs may be detected in other conditions with T-cell lymphopenia (DiGeorge syndrome, prematurity, certain syndromic conditions), so a low TREC is not diagnostic of SCID alone.
Confirmatory Diagnostic Workup
Diagnostic Criteria
A diagnosis of SCID is established when all three of the following are met:
- Absent or severely reduced T cells (CD3+ count < 300 cells/μL) with evidence of impaired T-cell function (PHA lymphocyte proliferation < 10% of control)
- No evidence of secondary cause of immunodeficiency (HIV — always exclude with HIV PCR; maternal medications; protein-losing enteropathy)
- Confirmed pathogenic mutation in a known SCID gene (or clinical phenotype consistent with SCID pending genetic confirmation)
Pathophysiology
SCID results from defects in genes critical for lymphocyte development, survival, or function. The common γ chain (γc, encoded by IL2RG) is shared by receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. IL-7 signalling is essential for thymic T-cell progenitor survival and expansion; IL-15 is essential for NK-cell development. Loss of γc therefore abolishes T and NK lymphopoiesis.
In ADA deficiency, the enzyme adenosine deaminase catalyses the deamination of adenosine to inosine and deoxyadenosine to deoxyinosine in the purine salvage pathway. Loss of ADA activity leads to accumulation of deoxyadenosine and its phosphorylated metabolite dATP, which:
- Inhibits ribonucleotide reductase, blocking DNA synthesis and lymphocyte proliferation
- Promotes apoptosis of developing thymocytes and peripheral lymphocytes
- Causes direct toxicity to hepatocytes, osteocytes, and auditory neurons through mitochondrial dysfunction
The degree of enzyme residual activity correlates with disease severity — complete absence causes classical infantile SCID; partial activity may present later (> 12 months) as a "leaky" or delayed-onset phenotype.
Immunological Consequences
All forms of SCID share a common functional outcome: absent adaptive immunity manifesting as:
- No T-cell–mediated immunity → no delayed-type hypersensitivity, no graft rejection, no control of viral/fungal/opportunistic infections
- Absent or non-functional B cells → no specific antibody production; immunoglobulin levels fall progressively as maternal IgG wanes
- Absent NK cells (in some forms) → impaired innate antiviral and anti-tumour surveillance
Investigations Summary
A tiered investigation strategy enables rapid presumptive diagnosis while genetic confirmation is awaited.
| Tier | Test | Expected in SCID | MBS Item |
|---|---|---|---|
| 1 — Immediate | FBC + differential | ALC < 3 × 10⁹/L; lymphocytes often < 1.5 × 10⁹/L | 65070 |
| 1 — Immediate | HIV PCR | Must be negative | 69497 |
| 1 — Immediate | Chest X-ray | Absent thymic shadow | 58506 |
| 2 — Urgent | Lymphocyte subsets (flow cytometry) | Phenotype-dependent (T−B+NK−, T−B−NK−, etc.) | 65090 |
| 2 — Urgent | Serum immunoglobulins | Low IgA, IgM; IgG declining | 65085 |
| 2 — Urgent | ADA enzyme activity (if T−B−NK−) | Absent / severely reduced | N/A (lab-specific) |
| 3 — Confirmatory | Genetic testing (panel/WES) | Pathogenic variant identified | 73288 |
| 3 — Confirmatory | TRECs / KRECs (if not done at NBS) | Absent/low TRECs | N/A (lab-specific) |
Risk Stratification & Prognosis
Transplant outcomes are heavily influenced by the infant's clinical condition at the time of HSCT. The following risk categories guide urgency and inform donor selection:
Treatment
Treatment of SCID has two phases: immediate stabilisation (bridging therapy) and definitive immune reconstitution (HSCT or gene therapy).
A. Bridging / Stabilisation Measures
Initiate immediately upon clinical suspicion — do not wait for genetic confirmation:
Supportive Measures
- Reverse isolation — single room, HEPA filtration, strict hand hygiene; minimise visitors; avoid soil/plant exposure
- Nutritional support — nasogastric or gastrostomy feeding if oral intake inadequate; dietitian involvement essential
- No live vaccines — BCG, rotavirus (RotaTeq/Rotarix), OPV, MMR, varicella, yellow fever, live attenuated influenza vaccine (LAIV) all contraindicated. Household contacts should not receive OPV or live attenuated vaccines that shed.
- Irradiated blood products — all transfusions must be irradiated and CMV-safe (leucodepleted) to prevent transfusion-associated GVHD and CMV transmission
- Genetic counselling — refer family to clinical genetics for carrier testing, prenatal diagnosis options, and reproductive planning
B. Definitive Treatment — Haematopoietic Stem Cell Transplantation (HSCT)
HSCT remains the gold standard and most widely available curative therapy for SCID in Australia.
Donor Priority
| Donor Category | Match | Expected Survival | Notes |
|---|---|---|---|
| Matched sibling donor (MSD) | HLA 10/10 | > 95% (pre-symptomatic) | Best outcome; available in ~25% of families |
| Matched unrelated donor (MUD) | HLA 10/10 or 9/10 | 85–95% | Search via Bone Marrow Donor Worldwide (BMDW) and Australian Bone Marrow Donor Registry (ABMDR) |
| Haploidentical parent | HLA 5/10 | 70–85% | Always available; T-cell depleted graft; faster immune reconstitution with post-transplant cyclophosphamide protocols |
| Umbilical cord blood (UCB) | Variable | 75–85% | Stored at Australian public cord blood banks; higher TRM, slower engraftment |
Conditioning Regimens
- T−B−NK− SCID (e.g. ADA-SCID, JAK3): Reduced-intensity conditioning (RIC) usually required — busulfan (targeted AUC-based dosing) + fludarabine ± serotherapy (anti-thymocyte globulin or alemtuzumab). Avoid irradiation in radiosensitive SCID (Artemis/DCLRE1C).
- T−B+ NK− SCID (e.g. X-linked SCID): Reduced-intensity or no conditioning; NK-cell alloreactivity may assist engraftment. Some centres use no conditioning for MSD transplants with excellent results.
- Artemis/DCLRE1C deficiency: Absolutely avoid total body irradiation and busulfan is used with caution — alkylator-free regimens with fludarabine + treosulfan preferred.
C. Definitive Treatment — Gene Therapy
Autologous gene therapy using lentiviral vector–mediated gene transfer has transformed the treatment landscape, particularly for ADA-SCID.
Gene Therapy vs HSCT — Key Considerations
- Advantages of gene therapy: No risk of GVHD (autologous cells); no need for immunosuppressive conditioning; avoids donor-related complications; available when no matched donor exists
- Limitations: Currently TGA/approved only for ADA-SCID; requires specialised manufacturing; insertional mutagenesis risk (very low with modern lentiviral vectors but long-term monitoring required); single-centre access in Australia
- Current status for X-linked SCID: Lentiviral gene therapy trials ongoing internationally (e.g. in the US via FDA IND); not yet standard of care in Australia. HSCT remains first-line for IL2RG-deficient SCID.
D. Enzyme Replacement Therapy (ERT) — PEG-ADA
Monitoring
Pre-Transplant Monitoring
- Weekly FBC, CRP, liver function tests
- CMV, adenovirus, EBV viral load (PCR) — weekly from diagnosis
- Respiratory virus PCR panel if symptomatic
- Growth monitoring — weight, length, head circumference fortnightly on WHO centile charts
- IgG trough levels before each IVIg infusion — target > 8 g/L
Post-Transplant Monitoring
- Chimerism studies (STR or donor-specific PCR) — day +30, +60, +100, +180, +365, then annually
- Lymphocyte subsets (CD3, CD4, CD8, CD19, CD16/56) — monthly for first 6 months, then 3-monthly
- IgG levels and immunoglobulin subclass — continue IVIg until B-cell engraftment and specific antibody responses confirmed
- Vaccine responses — check post-immunisation serology (tetanus, Hib, pneumococcal) at 12 and 24 months post-HSCT
- CMV, EBV, adenovirus monitoring — weekly during first 100 days, then as clinically indicated
- GVHD surveillance — skin, gut, liver assessment at each visit
- Neurodevelopmental assessment — particularly in ADA-SCID; early intervention referral if delay identified
Long-Term Follow-Up
- Annual immunology review — lifelong at a specialist centre
- Catch-up vaccination programme — live vaccines only after confirmed immune reconstitution (typically > 12–24 months post-HSCT, with specialist clearance)
- Fertility counselling — conditioning may affect fertility; refer to reproductive endocrinology
- Secondary malignancy surveillance — particularly with alkylating conditioning regimens
Special Populations
Empirical Therapy for Acute Infections
Any febrile episode in an infant with known or suspected SCID must be treated as a medical emergency. Initiate empirical broad-spectrum antimicrobials immediately after blood cultures and relevant specimens are obtained.
| Clinical Scenario | Empirical Regimen | Key Notes |
|---|---|---|
| Febrile neutropaenia / sepsis | Piperacillin-tazobactam 90 mg/kg (piperacillin component) IV 6-hourly ± gentamicin 7.5 mg/kg IV daily | Add vancomycin if central line infection suspected (15 mg/kg IV 6-hourly, dose-adjust per trough/AUC). Escalate to meropenem 40 mg/kg IV 8-hourly if no response in 48–72 hours. |
| Suspected PJP pneumonia | Co-trimoxazole 20 mg/kg/day (trimethoprim component) IV, divided TDS | High-dose for treatment (vs prophylactic dosing). Consider adjunctive corticosteroids if PaO₂ < 70 mmHg or A-a gradient > 35 mmHg. |
| Disseminated BCG-osis | Isoniazid 10–15 mg/kg/day PO + rifampicin 10–20 mg/kg/day PO + ethambutol 15–20 mg/kg/day PO (if age > 1 month, adjusted) ± amikacin 15 mg/kg IV daily (2–4 weeks) | BCG is a live attenuated vaccine — disseminated disease is life-threatening in SCID. Refer to RHDAustralia and infectious disease specialist. Expedite definitive therapy (HSCT/gene therapy). |
| Invasive candidiasis | Caspofungin 25 mg/m² IV loading then 50 mg/m²/day OR liposomal amphotericin B 3–5 mg/kg IV daily | Remove central lines if possible. Ophthalmology review. Minimum 14 days after first negative blood culture. |
| CMV viraemia / disease | Ganciclovir 5 mg/kg IV 12-hourly (induction) then 5 mg/kg IV daily (maintenance) | Monitor FBC closely (neutropaenia). Transition to valganciclovir PO when clinically stable. Letermovir increasingly used as prophylaxis post-HSCT. |
Aboriginal and Torres Strait Islander Health Considerations
SCID occurs in Aboriginal and Torres Strait Islander communities and requires culturally safe, equity-focused care. Several unique considerations apply:
📚 References
- 1. Kwan A, Abraham RS, Bhatt D, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738.
- 2. Heimall J, Logan BR, Cowan MJ, et al. Survival outcomes among patients with severe combined immunodeficiency who received transplants. JAMA Pediatr. 2018;172(5):e180737.
- 3. Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371(5):434–446.
- 4. Cicalese MP, Ferrua F, Castagnaro L, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128(1):45–54.
- 5. Gaspar HB, Cooray S, Gilmour KC, et al. Hematopoietic stem cell gene therapy for adenosine deaminase–deficient SCID. N Engl J Med. 2009;360(5):447–458.
- 6. Australian Government Department of Health and Aged Care. Australian National Immunisation Programme — Immunisation Handbook. Canberra: Australian Government; 2024. Available from: https://immunisationhandbook.health.gov.au
- 7. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW; 2023.
- 8. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S182–S194.
- 9. Chinn IK, Chan AY, Bhatt D, et al. Diagnostic interpretation of lymphocyte subsets and T-cell receptor excision circles in suspected immunodeficiency. J Allergy Clin Immunol Pract. 2021;9(8):3085–3097.
- 10. RHDAustralia (NACCHO, ASID, CARPA). Management of Tuberculosis — CARPA Standard Treatment Manual. 8th ed. Alice Springs: RHDAustralia; 2022.
- 11. Australian Bone Marrow Donor Registry (ABMDR). Annual Report 2023. Sydney: ABMDR; 2023. Available from: https://www.abmdr.org.au
- 12. Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with X-linked severe combined immunodeficiency. N Engl J Med. 2019;380(16):1525–1534.
- 13. ACSQHC (Australian Commission on Safety and Quality in Health Care). National Safety and Quality Health Service Standards. 2nd ed. Sydney: ACSQHC; 2021.
- 14. Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129(3):597–604.