📋 Key Information Summary
- Approximately 5–10% of all cancers are attributable to inherited germline mutations in high-penetrance tumour-suppressor or DNA-repair genes.
- The three most clinically significant familial cancer syndromes in Australian practice are Hereditary Breast and Ovarian Cancer (HBOC; BRCA1/2), Lynch Syndrome (MLH1/MSH2/MSH6/PMS2), and Familial Adenomatous Polyposis (FAP; APC).
- Refer for genetic assessment when there is a known pathogenic variant in the family, cluster of cancers meeting Manchester/modified Amsterdam criteria, early-onset disease, bilateral or multifocal tumours, or rare cancer–associated pathology.
- Medicare Benefits Schedule (MBS) items 73298–73304 fund germline genetic testing through approved Australian genetics laboratories when criteria are met.
- Pre-test genetic counselling is mandatory; informed consent must address implications for the individual and at-risk blood relatives.
- Cascade testing of first-degree relatives of a confirmed carrier is the most cost-effective population-level intervention and should be offered promptly.
- Risk-reducing bilateral salpingo-oophorectomy (RRBSO) in BRCA1 carriers reduces ovarian cancer mortality by ~80% and is recommended by age 35–40 for BRCA1 and 40–45 for BRCA2.
- Risk-reducing mastectomy (RRM) lowers breast cancer risk by ≥90% but is not mandatory; MRI-based surveillance is an accepted alternative per Cancer Australia guidelines.
- For Lynch Syndrome, annual colonoscopy should commence at age 20–25 (or 2–5 years before the earliest CRC diagnosis in the family) and continue every 1–2 years.
- Aspirin 600 mg daily for ≥2 years reduces Lynch-associated colorectal cancer incidence by ~63% (CAPP2 trial); discuss dose, duration, and GI bleeding risk.
- FAP requires surveillance flexible sigmoidoscopy/colonoscopy from age 10–12; prophylactic colectomy is indicated when polyp burden becomes unmanageable, typically by late teens to mid-twenties.
- All genetic testing results must be disclosed within a multidisciplinary framework; variants of uncertain significance (VUS) should not guide clinical management.
- Aboriginal and Torres Strait Islander peoples face significant barriers to genetic services including geographic isolation, lower referral rates, and culturally unsafe models of care; proactive outreach through Aboriginal Medical Services is essential.
- eviQ Cancer Genetics protocols (eviQ.org.au) provide point-of-care Australian treatment and surveillance regimens endorsed by Cancer Institute NSW.
- Psychosocial support should be integrated at every stage; genetic diagnosis can cause significant anxiety, altered family dynamics, and survivor guilt.
Introduction & Australian Epidemiology
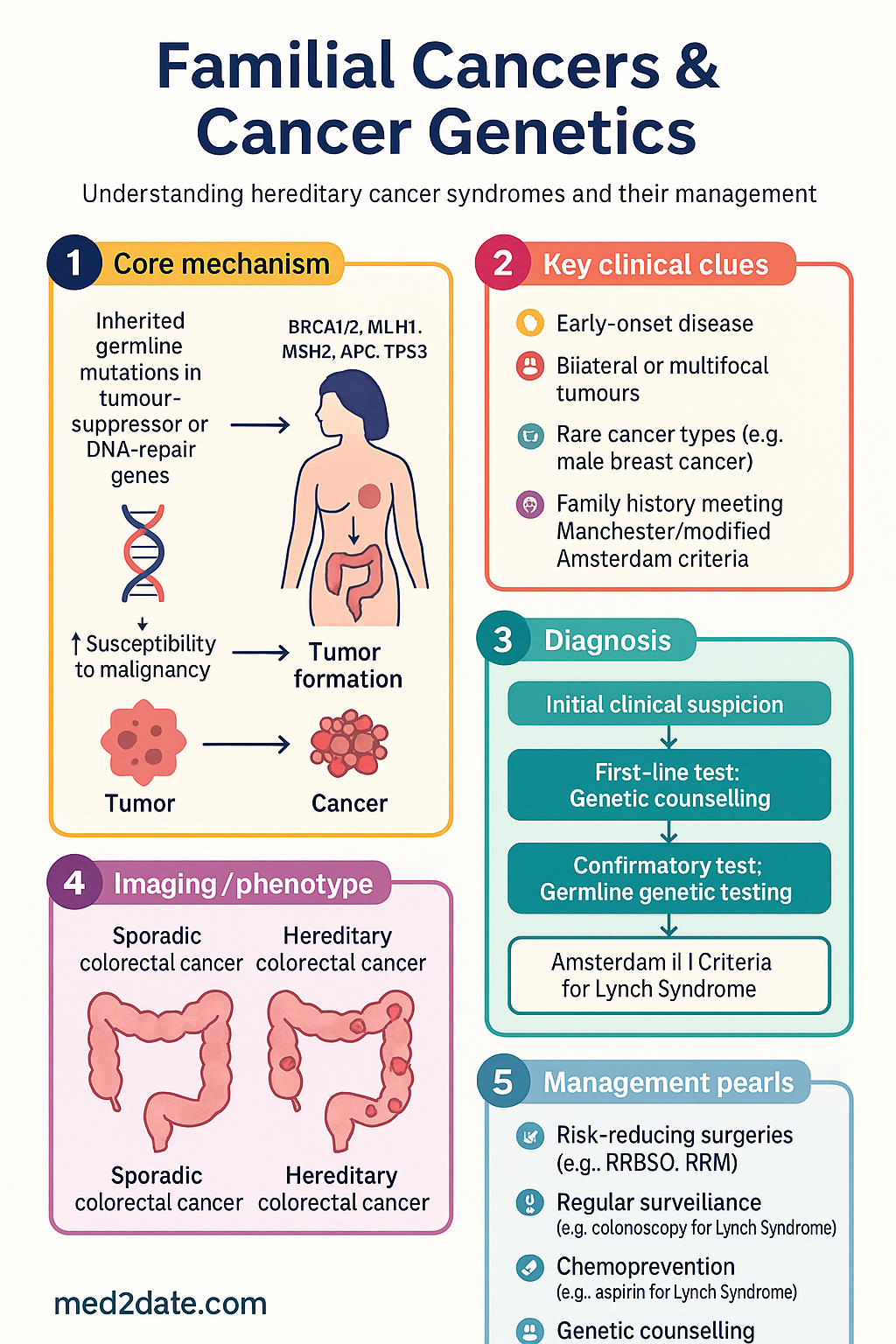
Familial cancers arise from inherited germline mutations in tumour-suppressor genes, DNA-mismatch-repair genes, or other cancer-predisposition loci. These mutations are present in every cell of the body from conception and confer a substantially elevated lifetime risk of one or more malignancies compared with the general population. In Australia, approximately 5–10% of all cancer diagnoses have a monogenic hereditary basis, and a further 15–20% show familial clustering suggestive of polygenic or shared environmental risk.
The identification of individuals carrying pathogenic germline variants has transformed cancer prevention, enabling tailored surveillance protocols, risk-reducing surgical interventions, and—in select cases—chemoprevention. The Australian landscape is well served by public Clinical Genetics Services in every state and territory, a national network of familial cancer centres, and publicly funded genetic testing via the Medicare Benefits Schedule (MBS).
In 2023, an estimated 165,000 Australians were diagnosed with cancer (AIHW). Of these, roughly 8,000–16,000 cases involved a hereditary component. BRCA1 and BRCA2 mutations alone account for approximately 5% of all breast cancers and 15–20% of all ovarian cancers nationally. Lynch Syndrome underlies 2–5% of all colorectal cancers, and APC-associated familial adenomatous polyposis accounts for <1% but carries near-100% penetrance for CRC without intervention.
This guideline provides an evidence-based framework for recognising, testing, counselling, surveilling, and managing individuals and families affected by hereditary cancer predisposition syndromes in the Australian context. It aligns with Cancer Australia, Human Genetics Society of Australasia (HGSA), eviQ, and international consensus standards.
Hereditary Syndromes Overview
Hereditary cancer syndromes are defined by the inheritance of a single high-penetrance pathogenic variant that dramatically increases lifetime cancer risk. Each syndrome typically involves a characteristic spectrum of tumour types, an earlier age of onset than sporadic counterparts, and frequently bilateral or multifocal disease. Recognition of the syndrome pattern is the critical first step in referral for genetic assessment.
Key Hereditary Cancer Syndromes in Australia
| Syndrome | Gene(s) | Primary Cancers | Lifetime Risk | Inheritance |
|---|---|---|---|---|
| HBOC | BRCA1, BRCA2 | Breast, ovarian, prostate, pancreatic | Breast 60–72% (BRCA1); 45–69% (BRCA2); Ovarian 39–44% (BRCA1); 11–17% (BRCA2) | AD |
| Lynch Syndrome | MLH1, MSH2, MSH6, PMS2, EPCAM | Colorectal, endometrial, ovarian, gastric, urinary tract, small bowel, hepatobiliary | CRC 52–82% (MLH1/MSH2); Endometrial 25–60% | AD |
| FAP | APC | Colorectal (hundreds–thousands of adenomas), duodenal, thyroid (papillary), hepatoblastoma | CRC ~100% without colectomy | AD |
| Li-Fraumeni | TP53 | Sarcoma, breast, brain, adrenocortical, leukaemia | Cancer >50% by age 30; >90% lifetime | AD |
| MEN1 | MEN1 | Parathyroid, pituitary, pancreatic neuroendocrine | >95% penetrance by age 50 | AD |
| MEN2 | RET | Medullary thyroid, phaeochromocytoma, parathyroid | MTC ~95% (MEN2A/2B) | AD |
| Cowden / PTEN-Hamartoma | PTEN | Breast, thyroid (follicular/papillary), endometrial | Breast 67–85%; Thyroid 35% | AD |
| Peutz-Jeghers | STK11 | GI (polyposis), breast, ovarian (sex-cord tumour), pancreatic | CRC 39–57%; Breast 31–54% | AD |
| VHL | VHL | Renal cell carcinoma, haemangioblastoma (CNS), phaeochromocytoma | >90% penetrance by age 65 | AD |
Referral Criteria for Clinical Genetics / Familial Cancer Centre
- Known pathogenic variant in the family (any cancer-predisposition gene).
- Individual cancer meeting age/family-number thresholds per Cancer Australia or eviQ criteria.
- Tumour molecular screening result indicating high probability of underlying germline variant (e.g., MMR-deficient / MSI-H CRC or endometrial carcinoma).
- Two or more relatives on the same side of the family with cancers consistent with a known syndrome.
- Ashkenazi Jewish ancestry with any personal or family history of breast or ovarian cancer (founder mutation frequency ~1 in 40).
Common Genes (BRCA1/2, APC, MLH1)
BRCA1 and BRCA2
BRCA1 (chromosome 17q21) and BRCA2 (chromosome 13q12.3) encode proteins essential for homologous recombination repair of double-strand DNA breaks. Pathogenic variants are inherited in an autosomal dominant pattern with high penetrance but variable expressivity.
- BRCA1: Lifetime breast cancer risk 60–72%; ovarian cancer risk 39–44%. BRCA1-associated breast cancers are frequently triple-negative (ER−/PR−/HER2−), high-grade, and diagnosed at a younger age.
- BRCA2: Lifetime breast cancer risk 45–69% (female); ovarian cancer risk 11–17%. Also confers elevated prostate cancer risk (up to 27% lifetime vs. ~12% general population) and pancreatic cancer risk (~5%). Male breast cancer risk is 6–8%.
- Australian founder mutations: Three Ashkenazi Jewish founder mutations (185delAG, 5382insC in BRCA1; 6174delT in BRCA2) account for a disproportionate share of BRCA-positive individuals in some Australian populations.
APC (Adenomatous Polyposis Coli)
APC is a tumour-suppressor gene on chromosome 5q22.2 and is the gatekeeper of the Wnt/β-catenin signalling pathway. Germline APC mutations cause Familial Adenomatous Polyposis (FAP), characterised by the development of hundreds to thousands of colorectal adenomas beginning in adolescence, with near-100% progression to colorectal cancer by the fifth decade if untreated.
- Classic FAP: >100 colorectal adenomas; CRC inevitable without colectomy.
- Attenuated FAP (AFAP): 10–100 adenomas, later onset; caused by mutations at the extreme 5′ or 3′ ends of APC or in the alternatively spliced exon 9.
- Genotype–phenotype correlation: Mutations between codons 1250–1464 are associated with the most profuse polyposis and earliest CRC onset. The 1309 codon hotspot carries particularly aggressive disease.
- Extracolonic manifestations: Duodenal adenomas/carcinomas (~5–12% lifetime risk), desmoid tumours (10–25%), congenital hypertrophy of the retinal pigment epithelium (CHRPE), papillary thyroid cancer, hepatoblastoma in children <5 years.
MLH1 and the Mismatch-Repair Genes
Lynch Syndrome (hereditary non-polyposis colorectal cancer, HNPCC) is caused by germline pathogenic variants in the DNA mismatch-repair (MMR) genes: MLH1, MSH2, MSH6, PMS2, or by deletions in the EPCAM gene causing epigenetic silencing of MSH2. The resultant MMR deficiency leads to accumulation of replication errors at microsatellite loci (microsatellite instability, MSI).
- MLH1 and MSH2 account for ~80–90% of diagnosed Lynch Syndrome in Australia; they carry the highest penetrance for CRC and extracolonic cancers.
- MSH6 mutations have lower CRC penetrance but higher endometrial cancer risk; later age of onset.
- PMS2 mutations are the least penetrant; some carriers may never develop cancer.
- Tumour screening: All colorectal and endometrial cancers in Australia should undergo immunohistochemistry (IHC) for MMR protein expression and/or MSI testing as per Cancer Australia and RACGP recommendations. Loss of MLH1 expression should prompt BRAF V600E mutation testing and/or MLH1 promoter methylation analysis to distinguish sporadic from hereditary loss.
- Amsterdam II Criteria (clinical): ≥3 relatives with Lynch-associated cancer, one first-degree relative of the other two; ≥2 successive generations affected; ≥1 diagnosed <50 years; FAP excluded.
- Bethesda Guidelines (molecular triage): CRC diagnosed <50 years; synchronous/metachronous Lynch-associated tumours; MSI-H histology in CRC <60 years; ≥1 first-degree relative with CRC <50 years; ≥2 first- or second-degree relatives with Lynch-associated tumours at any age.
Genetic Testing & Counselling
Pre-Test Counselling
All individuals referred for germline genetic testing must receive pre-test counselling from a qualified genetic counsellor or clinical geneticist. This may occur in person, by telehealth, or through an approved shared-care model. Pre-test counselling must cover:
- Nature and inheritance pattern of the suspected syndrome.
- Interpretation of possible results: pathogenic variant, likely pathogenic, variant of uncertain significance (VUS), likely benign, benign.
- Implications for life insurance, income protection, and superannuation (note: Australian moratorium on genetic testing in life insurance under 0,000, effective June 2019).
- Duty to inform at-risk relatives and familial disclosure support.
- Psychological impact and availability of support services (e.g., Cancer Council 13 11 20, Pink Hope, Lynch Syndrome Australia).
- Data privacy and storage of genetic information.
Genetic Testing Modalities in Australia
| Test Type | Technique | MBS Item | Indication | Turnaround |
|---|---|---|---|---|
| Single-gene testing | Sanger sequencing / targeted NGS | 73298 | Known familial pathogenic variant (cascade testing) | 2–4 weeks |
| Targeted gene panel | NGS panel (e.g., cancer predisposition panel 20–50 genes) | 73299–73301 | Clinical suspicion of hereditary cancer without known familial variant; tumour screening suggests Lynch Syndrome | 4–8 weeks |
| Large rearrangement analysis | MLPA / chromosomal microarray | 73302 | Deletion/duplication screening when sequencing is negative but clinical suspicion remains high | 3–6 weeks |
| Whole-exome / whole-genome sequencing | WES / WGS | 73303–73304 (restricted) | Panel-negative, high clinical suspicion; research or diagnostic odyssey | 8–16 weeks |
Post-Test Counselling & Variant Interpretation
- Pathogenic / Likely Pathogenic variant: Discuss penetrance, cancer risks, surveillance, and risk-reducing options. Issue a referral letter to the GP and relevant specialists. Offer cascade testing to at-risk relatives.
- Variant of Uncertain Significance (VUS): Do NOT alter clinical management based on a VUS. Advise re-contact when variant classification changes. Functional studies may be pursued in research settings.
- Benign / Likely Benign: Reassure. If clinical suspicion remains high, consider alternative genes or re-evaluate family history.
- Incidental / secondary findings: ACMG SF v3.2 list of medically actionable genes should be offered as an opt-in component of panel or exome testing.
Cascade Testing
Cascade testing—testing at-risk relatives for a known familial pathogenic variant—is the most efficient form of genetic testing and carries the highest positive predictive value. It is funded under MBS item 73298 and should be offered to all first-degree relatives of a confirmed carrier. Many Australian familial cancer centres operate dedicated cascade clinics and can facilitate testing by letter or through GP-initiated referrals.
Surveillance & Risk Reduction
Surveillance and risk-reducing strategies are syndrome-specific and must be individualised based on the gene involved, penetrance data, patient preference, comorbidities, and surgical fitness. The following evidence-based protocols are aligned with Cancer Australia, eviQ, and international guidelines.
Hereditary Breast and Ovarian Cancer (BRCA1/2)
Lynch Syndrome
| Organ | Surveillance | Start Age | Frequency | Notes |
|---|---|---|---|---|
| Colorectal | Colonoscopy | 20–25 years (or 2–5 years before earliest family CRC) | Every 1–2 years | Use high-definition colonoscopy with chromoendoscopy if available |
| Endometrial / Ovarian | Consider annual transvaginal ultrasound + endometrial biopsy; discuss risk-reducing hysterectomy ± BSO from age 35–40 | 30–35 years | Annual (if surveillance chosen) | Risk-reducing hysterectomy + BSO is the most effective gynaecological risk-reduction strategy |
| Gastric | Upper GI endoscopy | 30–35 years | Every 2–3 years | Higher priority for MSH2 and MLH1 carriers; H. pylori testing and eradication |
| Urinary tract | Urinalysis ± renal ultrasound | 30–35 years | Annual urinalysis | Primarily for MSH2 carriers |
Familial Adenomatous Polyposis (FAP)
- Colorectal surveillance: Flexible sigmoidoscopy or colonoscopy annually from age 10–12 years.
- Prophylactic colectomy: Indicated when polyp burden is unmanageable endoscopically. Options include total proctocolectomy with ileal pouch–anal anastomosis (IPAA) or total colectomy with ileorectal anastomosis (IRA). Timing: typically late teens to mid-twenties.
- Upper GI surveillance: OGD with duodenoscopy every 1–3 years from age 20–25 (Spigelman staging guides interval).
- Thyroid surveillance: Annual thyroid ultrasound from age 15–20.
- Hepatoblastoma screening: Serum alpha-fetoprotein (AFP) and liver ultrasound every 6 months from birth to age 5–7 in APC mutation carriers.
- Chemoprevention: Sulindac 150–200 mg PO BD reduces rectal polyp burden post-colectomy (not PBS-listed for this indication). Celecoxib 400 mg PO daily has demonstrated efficacy but carries cardiovascular risk.
Additional Syndromes
| Syndrome | Key Surveillance | Risk-Reducing Measures |
|---|---|---|
| Li-Fraumeni (TP53) | Whole-body MRI annually from birth; brain MRI annually; breast MRI annually from age 20 (female); abdominal USS/urinary catecholamines annually (children) | Avoid ionising radiation where possible (use MRI over CT); consider risk-reducing mastectomy |
| MEN1 | Calcium, PTH, prolactin, IGF-1 from age 5; MRI pituitary from age 5; CT/MRI pancreas from age 10; chromogranin A | Proton pump inhibitor for gastric acid hypersecretion; surgical parathyroidectomy for hyperparathyroidism |
| MEN2 (RET) | Calcitonin surveillance or prophylactic thyroidectomy (age dependent on RET codon: highest risk → within 6 months of life; high risk → by age 5; moderate risk → by age 5–10 or when calcitonin rises) | Prophylactic total thyroidectomy; annual plasma metanephrines from age 11 |
| VHL | Annual ophthalmic exam from age 1; annual MRI abdomen from age 11; annual MRI CNS/auditory from age 11; annual plasma metanephrines from age 5 | Nephron-sparing surgery for renal cell carcinoma <3 cm; stereotactic radiosurgery for CNS haemangioblastomas |
Pathophysiology
Hereditary cancer syndromes arise from germline mutations in genes that regulate cell growth, DNA repair, or apoptosis. Most follow the Knudson "two-hit" model: an individual inherits one inactivated allele (first hit) and somatic inactivation of the second allele (second hit) in a target tissue triggers clonal expansion and tumourigenesis. The specific gene and pathway disrupted determines the cancer spectrum and age of onset.
Key Pathways
- Homologous recombination repair (HRR): BRCA1 and BRCA2 are central to error-free repair of double-strand DNA breaks. Loss causes genomic instability and sensitivity to PARP inhibitors (synthetic lethality).
- DNA mismatch repair (MMR): MLH1, MSH2, MSH6, PMS2 correct single-base mismatches and insertion-deletion loops during replication. Deficiency causes microsatellite instability (MSI-H), neoantigen generation, and a pro-inflammatory tumour microenvironment.
- Wnt/β-catenin signalling: APC normally targets β-catenin for proteasomal degradation. APC loss stabilises β-catenin, constitutively activating Wnt target genes (MYC, CCND1) that drive adenoma formation.
- p53 tumour suppression: TP53 is the "guardian of the genome." Germline TP53 mutations disable cell-cycle arrest, apoptosis, and senescence, leading to multi-organ cancer predisposition at very young ages.
Clinical Presentation & Diagnostic Criteria
Hereditary cancer syndromes are most often identified through a combination of family history assessment, tumour molecular screening, and clinical diagnostic criteria. The following frameworks guide recognition and referral.
Family History Assessment Tools
- Three-generation pedigree: Essential for all patients presenting with cancer at any site. Document cancer types, ages of onset, bilaterality, consanguinity, and ethnic background.
- Manchester Scoring System: Assigns points for cancer types and ages; cumulative score ≥15 indicates ≥10% probability of carrying a BRCA1/2 mutation — refer to genetics.
- Tyrer-Cuzick (IBIS) model: Estimates 10-year and lifetime breast cancer risk incorporating family history, hormonal factors, and breast density. Scores ≥17% (threshold varies by guidelines) warrant MRI surveillance.
- Amsterdam II / revised Bethesda criteria: Guide referral and tumour screening for Lynch Syndrome.
Tumour-Based Screening (Somatic → Germline Cascade)
Universal tumour screening for Lynch Syndrome has been endorsed in Australia. All patients diagnosed with colorectal or endometrial cancer should undergo:
- Immunohistochemistry (IHC) for MLH1, MSH2, MSH6, PMS2 protein expression.
- MLH1 promoter methylation and/or BRAF V600E testing if MLH1 is absent (to exclude sporadic methylation).
- MSI testing (PCR-based or NGS-based) as an alternative or adjunct.
- Referral for germline genetic testing when abnormal screening is not explained by methylation/BRAF.
Investigations
Investigation in familial cancer genetics spans tumour-based screening (to triage for germline testing) and germline genetic testing itself. The following investigations are available through Australian public and private pathology services.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Psychosocial Support & Multidisciplinary Care
A genetic diagnosis of cancer predisposition can have profound psychosocial consequences including anxiety about future cancer risk, guilt about passing mutations to children, altered family dynamics, and the stress of ongoing surveillance. Integrated psychosocial support is a core component of familial cancer care.
- All patients should be offered psychological screening at diagnosis and at key decision points (e.g., pre-surgery, surveillance results).
- Referral pathways should include clinical psychology, psychiatry (where indicated), social work, and peer-support organisations (Pink Hope, Lynch Syndrome Australia, Cancer Council support groups).
- Multidisciplinary team (MDT) discussion should include clinical geneticist, genetic counsellor, relevant surgical and oncology specialists, and the patient's GP.
- Decision aids for risk-reducing surgery (e.g., Cancer Australia BRCA decision aid) support informed, values-based choice.
📚 References
- 1. Cancer Australia. Cancer risk management for women at high risk of breast/ovarian cancer — Clinical guidance. Surry Hills, NSW: Cancer Australia; 2024. Available from: canceraustralia.gov.au.
- 2. eviQ Cancer Genetics. Risk management for a person with a BRCA1 or BRCA2 mutation. Cancer Institute NSW; 2024. Protocol ID: 1579. Available from: eviq.org.au.
- 3. Human Genetics Society of Australasia (HGSA). Position Statement: Genetic testing and screening in childhood. HGSA; 2023.
- 4. Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855–1863.
- 5. Australian Institute of Health and Welfare (AIHW). Cancer in Australia: In brief 2023. Cancer series no. 139. Cat. no. CAN 140. Canberra: AIHW; 2023.
- 6. Provenzale D, Gupta S, Ahnen DJ, et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2016;14(8):1010–1030.
- 7. Gargiulo S, Reynolds BA, Engel JA, et al. Genetic counselling and testing for hereditary cancer predisposition in Australia: current practice and future directions. Med J Aust. 2022;217(4):185–188.
- 8. National Health and Medical Research Council (NHMRC). Ethical aspects of human genetic testing: An information paper. Canberra: NHMRC; 2020.
- 9. Cancer Council Australia Colorectal Cancer Guidelines Working Party. Clinical practice guidelines for the prevention, early detection and management of colorectal cancer. Sydney: Cancer Council Australia; 2017. Available from: wiki.cancer.org.au.
- 10. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–2416.
- 11. Royal Australian College of General Practitioners (RACGP). Guidelines for preventive activities in general practice. 10th ed. East Melbourne: RACGP; 2024.
- 12. Lindor NM, McMaster ML, Lindor CJ, Greene MH. Concise handbook of familial cancer susceptibility syndromes. J Natl Cancer Inst Monogr. 2008;(38):1–93.
- 13. Medicare Benefits Schedule (MBS). Category 6 — Pathology, Subgroup 13 — Genetic testing. Australian Government Department of Health; 2024. Available from: mbsonline.gov.au.
- 14. Cancer Australia. National Aboriginal and Torres Strait Islander Cancer Framework. Surry Hills, NSW: Cancer Australia; 2015.
- 15. Randall-David E, Hartman A, Bartsch R, et al. Access to genetic counselling for Indigenous Australians: a systematic review. Aust N Z J Public Health. 2023;47(2):100053.