📋 Key Information Summary
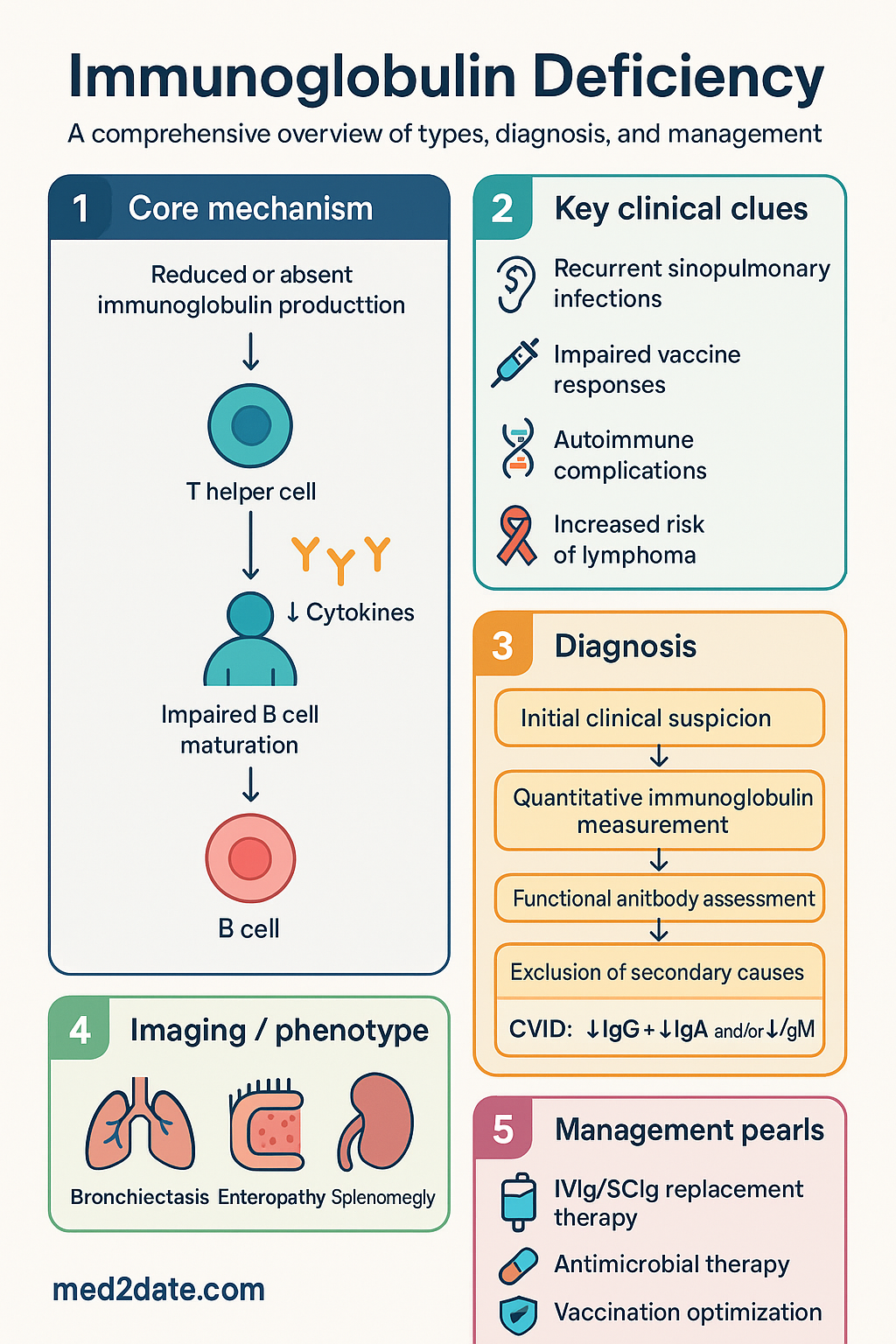
- Immunoglobulin (Ig) deficiency encompasses selective IgA deficiency, common variable immunodeficiency (CVID), IgG subclass deficiencies, and selective IgM deficiency — all predispose to recurrent sinopulmonary and mucosal infections.
- Selective IgA deficiency (IgA <0.07 g/L with normal IgG/IgM) is the most prevalent primary immunodeficiency worldwide, affecting approximately 1 in 300–700 Caucasians; prevalence in Aboriginal and Torres Strait Islander populations is poorly characterised.
- CVID is the most common symptomatic primary immunodeficiency in adults, characterised by hypogammaglobulinaemia, impaired vaccine responses, and increased susceptibility to autoimmune disease, granulomatous inflammation, and lymphoproliferative malignancy.
- Diagnosis requires quantitative immunoglobulin measurement (IgG, IgA, IgM, IgE), functional antibody assessment (anti-pneumococcal, anti-tetanus), lymphocyte subset analysis, and exclusion of secondary causes (medications, malignancy, nephrotic syndrome, protein-losing enteropathy).
- Intravenous immunoglobulin (IVIg) or subcutaneous immunoglobulin (SCIg) replacement is the cornerstone of management for CVID and symptomatic IgG subclass deficiency; Australian Government–funded IVIg/SCIg access requires State/Territory Blood Authority approval.
- Selective IgA deficiency rarely requires immunoglobulin replacement; most patients are managed with prompt antimicrobial therapy, vaccination optimisation, and mucosal hygiene measures.
- IgA-deficient patients receiving blood products must receive IgA-depleted plasma to prevent anaphylaxis from pre-formed anti-IgA antibodies.
- Pneumococcal vaccination with 13-valent conjugate (Prevenar 13®) followed by 23-valent polysaccharide (Pneumovax 23®) is essential; vaccine response assessment guides further management decisions.
- Empirical antibiotics for acute infective exacerbations: amoxicillin–clavulanate first-line for lower respiratory tract infection; trimethoprim–sulfamethoxazole or doxycycline for Pneumocystis jirovecii prophylaxis when CD4 counts are reduced.
- Aboriginal and Torres Strait Islander peoples experience higher rates of respiratory infections, bronchiectasis, and chronic suppurative lung disease — immunoglobulin deficiency should be considered in those with recurrent or severe sinopulmonary infections.
- Long-term complications include progressive bronchiectasis, enteropathy, splenomegaly, autoimmune cytopenias, and a 1.8- to 4-fold increased risk of lymphoma; structured annual review is mandatory.
- SCIg is increasingly preferred over IVIg for maintenance therapy in Australia, offering fewer systemic adverse effects, self-administration capability, and reduced hospital attendance burden — particularly advantageous for rural and remote patients.
Introduction & Australian Epidemiology
Immunoglobulin deficiency encompasses a heterogeneous group of primary and secondary immune disorders characterised by reduced or absent immunoglobulin production, predisposing affected individuals to recurrent sinopulmonary infections, mucosal infections, and immune dysregulation syndromes. Primary immunodeficiencies (PIDs) are intrinsic defects in immune cell development or function, whereas secondary causes — including nephrotic syndrome, protein-losing enteropathy, immunosuppressive therapy, haematological malignancy, and anti-convulsant medications — are more prevalent in general practice and must be excluded during diagnostic evaluation.
In Australia, selective IgA deficiency is estimated to affect 1 in 300–700 individuals of European descent, though significant underdiagnosis likely exists because most patients remain asymptomatic. The Australian Society of Clinical Immunology and Allergy (ASCIA) register records approximately 1,500–2,000 patients with CVID nationally, with an incidence of approximately 1 per 25,000–50,000. CVID typically presents in the second to fourth decade of life, with a slight female predominance in some cohorts. Secondary immunoglobulin deficiency is considerably more common, particularly in patients receiving rituximab, mycophenolate, or high-dose corticosteroids, and in those with nephrotic syndrome or B-cell lymphoproliferative disorders.
The burden of sinopulmonary disease in immunoglobulin-deficient patients is substantial. A 2019 Australian audit demonstrated that over 60% of CVID patients had radiographic evidence of bronchiectasis at diagnosis, and that delayed diagnosis — averaging 5–7 years after symptom onset — was associated with significantly worse pulmonary outcomes. Aboriginal and Torres Strait Islander peoples, who already experience disproportionately high rates of bronchiectasis and chronic suppurative lung disease, may be further disadvantaged if underlying immunoglobulin deficiency is not considered in the differential diagnosis.
This guideline provides a structured approach to the classification, diagnosis, and management of immunoglobulin deficiency in the Australian healthcare context, with emphasis on PBS-listed therapeutics, Australian Blood Authority–funded immunoglobulin access pathways, and considerations for equity of care in rural, remote, and Aboriginal and Torres Strait Islander communities.
Types of Immunoglobulin Deficiency
Selective IgA Deficiency
Selective IgA deficiency (SIgAD) is defined as a serum IgA level below 0.07 g/L in patients aged >4 years, with normal IgG and IgM concentrations and no other identified cause of hypogammaglobulinaemia. It is the most common primary immunodeficiency in Western populations. Most individuals are asymptomatic, but approximately 10–15% develop clinically significant recurrent respiratory infections, gastrointestinal disease, or autoimmune disorders. Patients who develop anti-IgA antibodies (approximately 1 in 300 SIgAD patients) are at risk of anaphylaxis during transfusion of IgA-containing blood products.
Common Variable Immunodeficiency (CVID)
CVID is the most prevalent symptomatic primary immunodeficiency in adults and is diagnosed when all of the following criteria are met: (a) markedly reduced IgG (at least 2 SD below age-adjusted mean) plus reduced IgA and/or IgM; (b) absent or markedly impaired vaccine responses; (c) exclusion of secondary causes; (d) onset typically after age 2 years. The European Society for Immunodeficiencies (ESID) and Pan-American Group for Immunodeficiency (PAGID) criteria are the international diagnostic standard. CVID is clinically heterogeneous, with approximately 70% presenting with infections alone, and 30% manifesting immune dysregulation — including autoimmune cytopenias, granulomatous-lymphocytic interstitial lung disease (GLILD), enteropathy, and splenomegaly. Genetic mutations in TNFRSF13B (TACI), ICOS, BAFF-R, and LRBA are identified in approximately 10–20% of patients.
IgG Subclass Deficiency
IgG subclass deficiency refers to isolated reduction in one or more IgG subclasses (IgG1, IgG2, IgG3, or IgG4) with a normal total IgG level. IgG1 deficiency accounts for approximately 65% of IgG in serum; IgG2 deficiency is most commonly associated with impaired responses to polysaccharide antigens. Clinically significant IgG subclass deficiency should only be diagnosed in the presence of recurrent infections and impaired functional antibody responses — isolated subclass reductions without clinical correlation do not warrant immunoglobulin replacement therapy.
Selective IgM Deficiency
Selective IgM deficiency (SIgMD) is defined as an isolated reduction in serum IgM below the age-adjusted normal range with normal IgG and IgA levels. It is an underrecognised entity with an estimated prevalence of 0.03–1.7%. SIgMD is associated with recurrent sinopulmonary infections, severe allergic disease, and autoimmune disorders. Management is directed at infection prevention; immunoglobulin replacement is rarely indicated.
| Type | Defining Feature | Prevalence | Infection Pattern |
|---|---|---|---|
| Selective IgA deficiency | IgA <0.07 g/L; normal IgG/IgM | 1 in 300–700 | Mucosal (respiratory, GI); most asymptomatic |
| CVID | ↓IgG + ↓IgA and/or ↓IgM; impaired vaccine responses | 1 in 25,000–50,000 | Sinopulmonary; encapsulated organisms; opportunistic if CD4↓ |
| IgG subclass deficiency | ↓1–2 IgG subclasses; normal total IgG | Variable (1–10% of referrals) | Recurrent URTI, sinusitis, otitis; polysaccharide non-responders |
| Selective IgM deficiency | ↓IgM; normal IgG/IgA | 0.03–1.7% | Sinopulmonary; encapsulated organisms |
Clinical Significance
The clinical significance of immunoglobulin deficiency varies substantially by type and severity. Key considerations include:
Infectious Complications
Patients with CVID and symptomatic IgG subclass deficiency suffer recurrent bacterial infections, most commonly involving the sinuses, middle ear, bronchi, and lungs. The predominant pathogens are encapsulated organisms — Streptococcus pneumoniae, Haemophilus influenzae (non-typeable), and Moraxella catarrhalis. Recurrent or severe infections lead to progressive bronchiectasis in up to 60% of CVID patients, which itself becomes a source of chronic infection with Pseudomonas aeruginosa and non-tuberculous mycobacteria. GI infections with Giardia lamblia, Campylobacter species, and norovirus are also overrepresented.
Autoimmune and Immune Dysregulation
Approximately 25–30% of CVID patients develop autoimmune complications, including autoimmune haemolytic anaemia (AIHA), immune thrombocytopenic purpura (ITP), autoimmune neutropenia, pernicious anaemia, and thyroiditis. Granulomatous-lymphocytic interstitial lung disease (GLILD) is a serious complication unique to CVID, presenting with diffuse pulmonary infiltrates and non-caseating granulomata, and is associated with significantly reduced survival.
Malignancy Risk
CVID carries a 1.8- to 4-fold increased risk of malignancy, predominantly non-Hodgkin lymphoma (particularly MALT lymphoma and diffuse large B-cell lymphoma) and gastric carcinoma. Regular surveillance — including age-appropriate cancer screening, gastroscopy for those with enteropathy, and low threshold for lymph node biopsy — is essential.
Quality of Life and Psychosocial Burden
Immunoglobulin deficiency significantly impairs quality of life. In a 2020 Australian patient survey, CVID patients reported a median of 14 days of illness per month, 3 hospitalisations in the preceding 12 months, and substantial impact on employment and social participation. The introduction of home-based SCIg therapy has demonstrated improvement in quality-of-life scores and reduced healthcare utilisation.
Diagnosis
The diagnostic approach to immunoglobulin deficiency requires a systematic evaluation combining quantitative immunoglobulin measurement, functional antibody assessment, exclusion of secondary causes, and — where indicated — advanced immunological testing. Diagnosis should be confirmed on at least two separate occasions at least 3 months apart.
Tier 1 — Initial Evaluation (Primary Care)
Tier 2 — Specialist Immunology Assessment
Tier 3 — Advanced / Genetic Testing
Management
Management of immunoglobulin deficiency encompasses immunoglobulin replacement therapy (when indicated), antimicrobial therapy for acute infections, vaccination optimisation, monitoring for complications, and patient education. The approach is tailored to the specific type and severity of deficiency.
Immunoglobulin Replacement Therapy
In Australia, access to government-funded immunoglobulin products is managed through the National Blood Authority (NBA) and State/Territory Blood Authorities. Indications for replacement are defined in the National Blood Authority Criteria for the Clinical Use of Immunoglobulin in Australia.
Antimicrobial Prophylaxis and Acute Therapy
Management by Immunoglobulin Type
Vaccination Strategies
Vaccination is a critical component of immunoglobulin deficiency management. However, vaccine responses are often impaired, necessitating specific strategies.
| Vaccine | Schedule | Notes |
|---|---|---|
| Pneumococcal conjugate (PCV13) | 1 dose as priming immunisation | Followed by PPV23 booster at ≥8 weeks; assess response to serotypes |
| Pneumococcal polysaccharide (PPV23) | 1 dose 8 weeks after PCV13; repeat every 5 years | Pre-vaccination serotype IgG drawn at time of vaccination; post-vaccination at 4–6 weeks |
| Influenza (inactivated) | Annual — high-dose or adjuvanted formulation preferred | Free under NIP for immunocompromised; live attenuated (FluMist®) contraindicated |
| COVID-19 | Additional doses per ATAGI guidance for immunocompromised | mRNA vaccines preferred; booster doses recommended at 6-month intervals |
| Haemophilus influenzae type b (Hib) | 2 primary doses + booster if not previously immunised or antibody-negative | Particularly important in CVID and IgG subclass deficiency |
Monitoring
Structured monitoring is essential to optimise immunoglobulin therapy, detect complications early, and improve long-term outcomes.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Aboriginal and Torres Strait Islander peoples experience a disproportionate burden of respiratory infections, bronchiectasis, and chronic suppurative lung disease compared to non-Indigenous Australians. Immunoglobulin deficiency — particularly CVID and IgG subclass deficiency — may be an underrecognised contributing factor in patients with recurrent or severe sinopulmonary infections. Culturally safe, equitable access to immunology specialist assessment and immunoglobulin replacement therapy is essential.
📚 References
- 1. National Blood Authority. National Policy: Criteria for the Clinical Use of Immunoglobulin in Australia. Canberra: NBA; 2023.
- 2. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary Immunodeficiency Position Statement. Sydney: ASCIA; 2023. Available at: https://www.allergy.org.au
- 3. Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) Registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. 2019;7(6):1763–1770.
- 4. Bogaert DJA, Dullaers M, Lambrecht BN, et al. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016;53(9):575–590.
- 5. Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematology Am Soc Hematol Educ Program. 2012;2012:301–305.
- 6. >resnov T, Milota T, Litzman J, et al. Global prevalence of common variable immunodeficiency: a systematic review and meta-analysis. Front Immunol. 2023;14:1181585.
- 7. Abolhassani H, Aghamohammadi A, Imai K, et al. TNFRSF13B (TACI) mutations in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2018;141(3):973–983.
- 8. AIHW (Australian Institute of Health and Welfare). Aboriginal and Torres Strait Islander Health Performance Framework 2020 Summary Report. Canberra: AIHW; 2020.
- 9. Chang AC, Gupte NL, Binks M, et al. Bronchiectasis in Indigenous children. Respirology. 2020;25(10):1044–1053.
- 10. Orange JS, Ballow M, Stiehm ER, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2012;130(3 Suppl):S1–S24.
- 11. Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59.
- 12. Queensland Health. Subcutaneous Immunoglobulin (SCIg) Home Therapy Programme — Clinical Guideline. Brisbane: Queensland Government; 2022.
- 13. American College of Rheumatology (ACR). 2022 Guideline for Vaccination in Patients with Rheumatic and Musculoskeletal Diseases. Arthritis Care Res. 2023;75(1):2–17.