📋 Key Information Summary
- IgG subclass deficiency (IgGSD) is a selective reduction in one or more IgG subclasses (IgG1–4) with normal total IgG, distinguishing it from common variable immunodeficiency (CVID).
- IgG2 deficiency is the most frequently identified subclass defect, particularly in paediatric populations, and is strongly associated with encapsulated organism infections.
- IgG3 deficiency is the second most common subclass deficiency in adults and often presents with recurrent upper and lower respiratory tract infections.
- IgG1 deficiency may occur in isolation but more commonly signals evolving CVID or other primary immunodeficiency; longitudinal surveillance is essential.
- IgG4 deficiency is rare and usually clinically insignificant; IgG4-related disease is a separate entity entirely.
- Diagnosis requires: (1) recurrent serious infections, (2) low age-adjusted subclass levels on at least two occasions ≥4 weeks apart, and (3) exclusion of secondary causes (protein loss, immunosuppression).
- Immunoglobulin replacement therapy (IgRT) with IVIg or SCIg is the primary treatment for patients with confirmed deficiency and significant infectious burden — this is PBS Authority Required.
- Vaccination response to polysaccharide and conjugate vaccines (especially Pneumovax® 23 and conjugate pneumococcal) is a functional assessment tool.
- First-line prophylactic antibiotics (e.g., amoxicillin or azithromycin) may be trialled before commencing IgRT, particularly in mild disease.
- Prognosis is generally favourable; some children — particularly those with IgG2 deficiency — may spontaneously resolve by adolescence.
- Aboriginal and Torres Strait Islander patients face higher infectious burdens and barriers to specialist access; earlier referral and community-based IgRT delivery should be considered.
- All patients require annual respiratory function testing, sputum culture surveillance, and HRCT chest if bronchiectasis is suspected.
Introduction & Australian Epidemiology
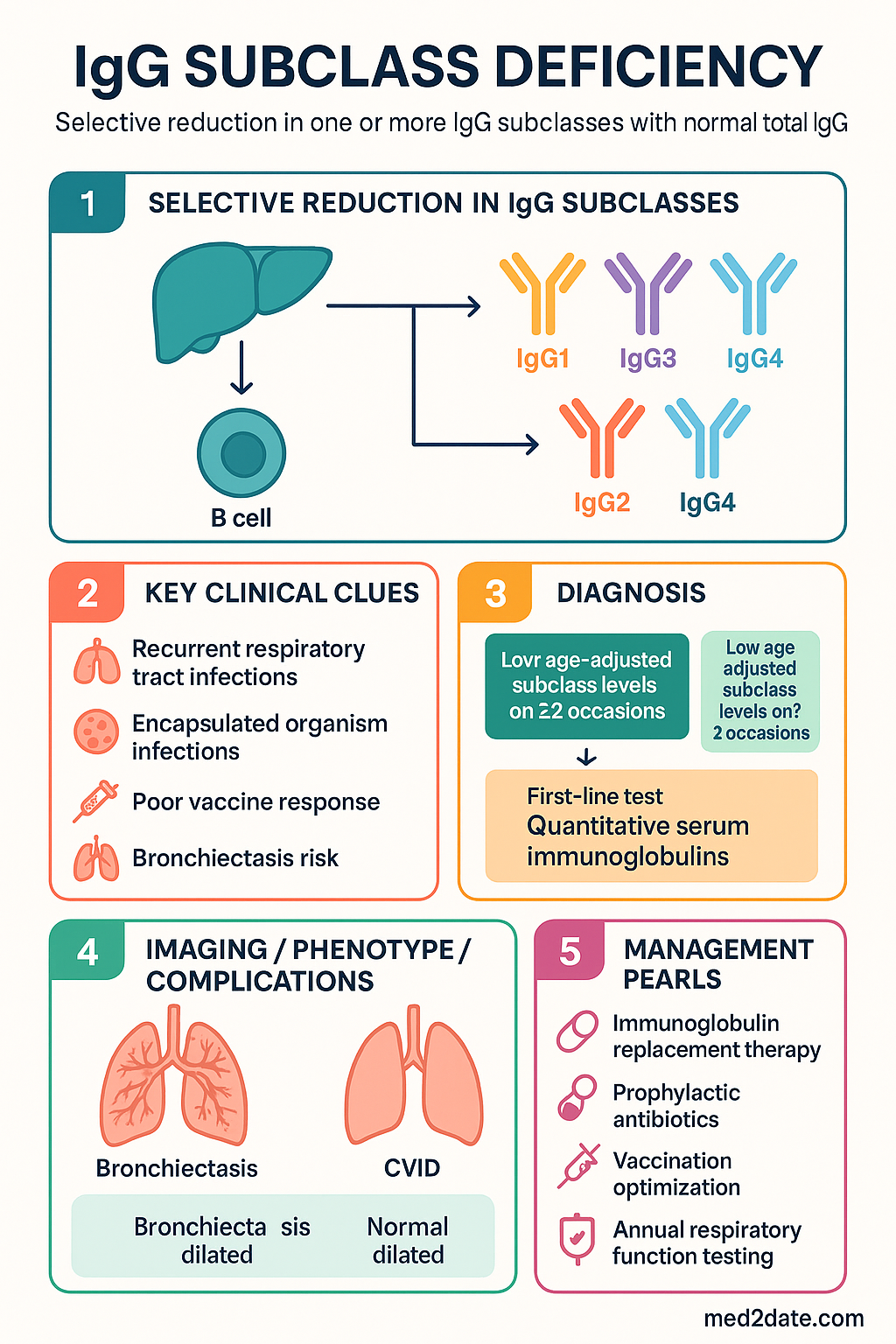
IgG subclass deficiency (IgGSD) is defined as a selective reduction in one or more of the four IgG subclasses (IgG1, IgG2, IgG3, or IgG4) below age- and laboratory-specific reference ranges, while total serum IgG remains within normal limits. It is classified among the predominantly antibody deficiencies in the International Union of Immunological Societies (IUIS) classification of inborn errors of immunity.
IgGSD sits on a spectrum of primary humoral immunodeficiency disorders. It must be distinguished from selective IgA deficiency, common variable immunodeficiency (CVID), and transient hypogammaglobulinaemia of infancy. The clinical significance of isolated subclass deficiency remains debated; many individuals with laboratory-defined IgGSD remain asymptomatic, while others develop serious recurrent infections, particularly of the respiratory tract.
In Australia, population-level prevalence data are limited, but IgG2 deficiency is estimated to affect 1 in 600–1,000 individuals, making it the most common subclass deficiency identified in both paediatric and adult immunology clinics. IgG3 deficiency is the most prevalent subclass deficiency in adult cohorts referred for recurrent infections. The Australian Paediatric Immunodeficiency Registry (APIR) captures cases in children, while adult data are largely extrapolated from tertiary centre series in Sydney, Melbourne, and Brisbane.
The economic and clinical burden is significant. Patients with IgGSD experience increased rates of community-acquired pneumonia, chronic sinusitis, otitis media, and — when undertreated — progressive bronchiectasis. Australian data from the Australasian Society of Clinical Immunology and Allergy (ASCIA) suggest that delays in diagnosis average 3–7 years from symptom onset, highlighting the need for increased awareness among general practitioners and respiratory physicians.
IgG Subclasses & Functions
IgG constitutes approximately 75% of total serum immunoglobulin and is divided into four subclasses based on heavy-chain constant-region structure. Each subclass has distinct effector functions, complement-activation capacity, and half-life.
| Subclass | % of Total IgG | Half-life (days) | Key Functions | Deficiency Associations |
|---|---|---|---|---|
| IgG1 | 60–70% | 21–23 | Protein antigen responses; complement activation (classical pathway); FcγR-mediated phagocytosis and ADCC | May signal evolving CVID; viral and bacterial infections |
| IgG2 | 15–25% | 21–23 | Polysaccharide antigen responses (encapsulated organisms); opsonisation; weak complement activator | Most common deficiency; recurrent sinusitis, pneumonia with Streptococcus pneumoniae, Haemophilus influenzae |
| IgG3 | 4–8% | 7–8 | Potent complement activator; antiviral responses; mucosal immunity | Second most common deficiency in adults; recurrent RTI, mucosal infections |
| IgG4 | 2–6% | 21–23 | Anti-inflammatory; blocks IgE-mediated reactions; does not activate complement | Usually clinically insignificant in isolation; do not confuse with IgG4-related disease |
IgG subclass levels vary with age. IgG1 reaches adult levels by age 5–7 years, IgG2 by age 8–10 years, IgG3 by age 10–12 years, and IgG4 is more variable. This is clinically important because IgG2 deficiency diagnosed in early childhood may spontaneously resolve as the immune system matures.
Polysaccharide responses depend critically on IgG2. Individuals with IgG2 deficiency typically fail to mount adequate IgG anti-capsular antibody to S. pneumoniae serotypes, predisposing them to invasive pneumococcal disease. The introduction of conjugate pneumococcal vaccines (7vPCV, 13vPCV) has partially mitigated this risk, but gaps remain for non-vaccine serotypes.
Clinical Features
The clinical presentation of IgGSD is heterogeneous, ranging from asymptomatic laboratory findings to severe recurrent infections with end-organ damage. The predominant clinical manifestation is recurrent respiratory tract infection, though the spectrum varies by subclass deficiency and patient age.
Symptoms & Signs
Subclass-Specific Presentations
- IgG2 deficiency: Predominantly encapsulated organism infections — otitis media with S. pneumoniae or H. influenzae, recurrent pneumonia, sinusitis. Particularly common in children <8 years. Associated with poor responses to unconjugated polysaccharide vaccines.
- IgG3 deficiency: Upper and lower RTIs in adults; may present with chronic cough, persistent sputum production, and recurrent bronchitis. Associated with atopic conditions in some studies.
- IgG1 deficiency: Broad-spectrum infection susceptibility; may overlap with early CVID. Patients with isolated IgG1 deficiency and significant infections should be monitored for progression to hypogammaglobulinaemia.
- IgG2 + IgG4 combined deficiency: Highest infectious burden; increased risk of bronchiectasis and invasive pneumococcal disease; often associated with poor vaccine responses.
Diagnosis
Diagnosis of IgGSD requires a systematic approach combining clinical assessment, quantitative immunoglobulin measurement, functional antibody evaluation, and exclusion of secondary causes. The diagnosis should be made or confirmed by a clinical immunologist or immunology-trained physician.
Diagnostic Criteria
- Clinical: Recurrent serious infections (particularly sinopulmonary), requiring antibiotics, with documented infections (microbiological confirmation preferred).
- Laboratory: One or more IgG subclass levels below the age- and sex-adjusted laboratory reference range on at least two occasions separated by ≥4 weeks.
- Total IgG: Within normal limits (distinguishes IgGSD from CVID or hypogammaglobulinaemia).
- Exclusion of secondary causes: Protein-losing enteropathy, nephrotic syndrome, immunosuppressive therapy, malignancy, viral suppression (HIV, EBV, CMV), and medication effects (carbamazepine, phenytoin, gold, sulfasalazine).
Investigations
Differential Diagnosis
| Condition | Distinguishing Features |
|---|---|
| Common Variable Immunodeficiency (CVID) | Total IgG, IgA, and/or IgG reduced; often presents in 2nd–3rd decade; higher infection burden; may have autoimmune or granulomatous complications |
| Transient Hypogammaglobulinaemia of Infancy | Low IgG for age (all subclasses may be proportionately reduced); self-resolving by age 2–4 years; normal antibody responses to vaccination |
| Selective IgA Deficiency | IgA <0.07 g/L with normal IgG and IgM; may coexist with IgG2/IgG4 deficiency; check before transfusing blood products (anti-IgA antibodies) |
| Specific Antibody Deficiency (SAD) | Normal IgG subclasses and total IgG but impaired polysaccharide antibody responses; clinical overlap with IgG2 deficiency |
| Secondary Immunodeficiency | Immunosuppressants, nephrotic syndrome, protein-losing enteropathy, malignancy, HIV, medications |
| Sinusitis/bronchitis without immunodeficiency | Anatomical factors, allergic rhinitis, gastro-oesophageal reflux, primary ciliary dyskinesia |
Management
Management of IgGSD follows a stepwise approach: (1) vaccination optimisation and infection prevention, (2) prophylactic antibiotics for moderate disease, and (3) immunoglobulin replacement therapy (IgRT) for severe or refractory disease. All patients should be co-managed with a clinical immunologist.
1. Vaccination Optimisation
- Pneumococcal vaccination: Administer 13-valent pneumococcal conjugate vaccine (13vPCV, Prevenar 13®) followed by Pneumovax® 23 at least 8 weeks later. Measure anti-pneumococcal IgG serotypes pre- and 4–6 weeks post-vaccination to assess functional response. Repeat Pneumovax® 23 every 5 years if antibody levels decline. Note: Pneumovax® 23 is funded under NIP for ATSI adults ≥50 years and all adults ≥70 years; other IgGSD patients require private prescription.
- Influenza vaccination: Annual quadrivalent influenza vaccination (NIP-funded) — standard dose; consider high-dose formulation for elderly patients.
- Other: Ensure up-to-date Hib (if incomplete), meningococcal ACWY and B, varicella (if non-immune and not on IgRT), and COVID-19 vaccination per ATAGI guidance.
- Live vaccines: Generally safe in isolated IgGSD (unlike XLA or severe combined immunodeficiency); however, defer live vaccines if the patient is on IgRT or has concurrent IgA deficiency, as anti-IgA antibodies may be present.
2. Prophylactic Antibiotics
For patients with moderate disease (frequent but non-severe infections) who do not yet meet criteria for IgRT, a trial of prophylactic antibiotics is appropriate:
3. Immunoglobulin Replacement Therapy (IgRT)
IgRT is indicated for patients with IgGSD who have:
- Documented serious or recurrent infections despite prophylactic antibiotics
- Evidence of bronchiectasis
- Documented functional antibody deficiency (poor vaccine responses) with significant infectious burden
- Hospitalisation for infection ≥1 in the preceding 12 months
Monitoring
- Trough IgG levels: Measure serum IgG trough (pre-infusion) at 3 months, 6 months, and then 6–12 monthly. Target trough ≥7 g/L or individualised to clinical response.
- Infection frequency: Document infection episodes, antibiotic use, hospitalisations, and quality of life at each visit.
- Spirometry: Annual FEV₁/FVC monitoring. Declining FEV₁ may indicate evolving bronchiectasis.
- HRCT chest: At baseline if bronchiectasis suspected; repeat only if clinical deterioration.
- Sputum surveillance: Annual sputum culture for patients with bronchiectasis or on macrolide prophylaxis.
- Renal function: Monitor serum creatinine before each IVIg infusion if using sucrose-containing preparations.
- Re-evaluation of subclass levels: Repeat IgG subclass panel annually, especially in paediatric patients, as spontaneous resolution may occur.
Management of IgRT Adverse Effects
| Adverse Effect | Management |
|---|---|
| Infusion-related reactions (headache, myalgia, chills) | Reduce infusion rate; pre-medicate with paracetamol, antihistamine ± hydrocortisone; ensure adequate hydration |
| Anaphylaxis (rare) | Stop infusion immediately; IM adrenaline; change brand or route (IVIg → SCIg); consider IgA-deficient preparations if concurrent IgA deficiency |
| Local site reactions (SCIg) | Rotate infusion sites; use hyaluronidase-facilitated SCIg (HyQvia®) if persistent |
| Haemolysis (IVIg) | Anti-A/anti-B haemolysin titre varies by brand; monitor FBC and haptoglobin; use low-titre preparations if recurrent |
| Thrombotic events (rare) | Slow infusion rate; avoid in patients with high viscosity risk; adequate hydration; consider antithrombotic prophylaxis if multiple risk factors |
Special Populations
Aboriginal and Torres Strait Islander Health
📚 References
- 1. Bousfiha A, Jeddane L, Picard C, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508–1520.
- 2. Orange JS, Ballow M, Stiehm ER, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the AAAAI. Clin Immunol. 2012;143(1):63–87.
- 3. Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205.
- 4. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary immunodeficiency diseases (PID) guide for clinicians. ASCIA; 2023. Available at: www.allergy.org.au.
- 5. Chapel H, Prevot J, Gaspar HB, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. Front Immunol. 2014;5:162.
- 6. Australian Technical Advisory Group on Immunisation (ATAGI). Australian Immunisation Handbook. Australian Government Department of Health; 2023. Available at: immunisationhandbook.health.gov.au.
- 7. Chang AC, Loh J, Tze WJ, et al. Immunodeficiency in Australia: a national audit of primary immunodeficiency diagnoses. Intern Med J. 2020;50(Suppl 4):28.
- 8. Jolliffe L, Chang AB, Boyd J, et al. Bronchiectasis in Indigenous Australian children: a narrative review. Med J Aust. 2021;215(9):425–430.
- 9. Cinetto F, Scarpa R, Rattazzi M, et al. The broad spectrum of inborn errors of immunity manifested with respiratory tract infections: an EAID survey. Front Immunol. 2021;12:653140.
- 10. PDfA (Poisons and Therapeutic Goods Administration). Australian Product Information — Intragam® P (human normal immunoglobulin). CSL Behring; 2023.
- 11. PDfA (Poisons and Therapeutic Goods Administration). Australian Product Information — Hizentra® (human normal immunoglobulin for subcutaneous use). CSL Behring; 2023.
- 12. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020.
- 13. Yong PF, Thaventhiran JE, Grimbacher B. "A rose is a rose is a rose," but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107.
- 14. Primary Immunodeficiency Foundation of Australia. Primary immunodeficiency in Australia: clinical guidelines. PIFA; 2022.