📋 Key Information Summary
- Humoral immunity is the antibody-mediated arm of adaptive immunity, providing defence against extracellular pathogens, toxins, and free viral particles via B cell–derived immunoglobulins.
- Naïve B cells are activated through T-dependent pathways (requiring CD4⁺ T follicular helper cells) or T-independent pathways (via direct polyclonal activation by TI-2 antigens such as polysaccharides).
- Following activation, B cells undergo clonal expansion, somatic hypermutation, and affinity maturation in germinal centres of secondary lymphoid organs.
- Antibodies consist of two heavy and two light chains forming Y-shaped glycoproteins with a variable antigen-binding fragment (Fab) and a constant crystallisable fragment (Fc) that mediates effector functions.
- Five immunoglobulin classes exist — IgG, IgA, IgM, IgD, and IgE — each with distinct tissue distribution, complement-activating capacity, and roles in immune defence.
- IgG is the most abundant serum immunoglobulin; it crosses the placenta (providing neonatal passive immunity), mediates opsonisation, and activates the classical complement pathway.
- IgA predominates at mucosal surfaces as secretory dimeric IgA (sIgA), neutralising pathogens at the portal of entry; deficiency is the most common primary immunodeficiency worldwide.
- IgM is the first antibody produced during a primary immune response; pentameric IgM is a potent activator of the classical complement cascade.
- The complement system comprises over 30 proteins activated via classical (antibody-dependent), lectin (mannose-binding lectin), or alternative (spontaneous C3 hydrolysis) pathways.
- Complement activation generates anaphylatoxins (C3a, C5a) for chemotaxis, opsonins (C3b) for phagocytosis, and the membrane attack complex (C5b–9) for direct pathogen lysis.
- Humoral immune deficiencies — including X-linked agammaglobulinaemia, common variable immunodeficiency (CVID), selective IgA deficiency, and complement deficiencies — present with recurrent sinopulmonary, encapsulated bacterial, and enteric infections.
- Australian clinical practice monitors humoral immunity via serum immunoglobulin quantification (MBS item 69440), specific antibody titres post-vaccination, complement levels (CH50, C3, C4), and flow cytometric B cell subset analysis.
Introduction & Australian Epidemiology
Humoral immunity is the B cell and antibody-mediated arm of adaptive immunity, providing protection against extracellular pathogens, toxins, and free viral particles. Unlike cell-mediated immunity, which relies on cytotoxic T lymphocytes to eliminate intracellularly infected cells, humoral immunity functions primarily in the extracellular space — neutralising toxins, opsonising bacteria for phagocytosis, activating the complement cascade, and facilitating antibody-dependent cellular cytotoxicity (ADCC).
The central effector molecules of humoral immunity are immunoglobulins (antibodies), which are secreted by terminally differentiated B cells known as plasma cells. The generation of a robust humoral response requires the coordinated interaction of B cells, CD4⁺ T follicular helper (Tfh) cells, dendritic cells, and follicular dendritic cells within the germinal centres of secondary lymphoid organs — principally lymph nodes, the spleen, and mucosa-associated lymphoid tissue (MALT).
Epidemiology of Humoral Immune Deficiency in Australia
Primary immunodeficiencies affecting humoral immunity are not rare in Australia. Selective IgA deficiency (sIgAD) affects approximately 1 in 300–700 individuals in Caucasian populations and is frequently underdiagnosed because many patients are asymptomatic. The Australasian Society of Clinical Immunology and Allergy (ASCIA) estimates that between 25,000 and 50,000 Australians live with clinically significant primary immunodeficiency, with antibody deficiencies constituting the largest category (~50 %).
Common variable immunodeficiency (CVID) has a prevalence of approximately 1 in 25,000. In Australia, the mean diagnostic delay for CVID is 5–7 years, during which patients may develop bronchiectasis, granulomatous disease, or autoimmune complications. X-linked agammaglobulinaemia (Bruton disease) is rarer (~1 in 100,000 male births) but presents in infancy with severe recurrent infections if intravenous or subcutaneous immunoglobulin replacement is not commenced promptly.
Acquired causes of impaired humoral immunity are also common. Chronic lymphocytic leukaemia (CLL), the most prevalent adult leukaemia in Australia (~1,400 new diagnoses annually per AIHW data), frequently causes hypogammaglobulinaemia through disruption of normal B cell development. Rituximab, an anti-CD20 monoclonal antibody widely prescribed for rheumatoid arthritis, ANCA-associated vasculitis, and haematological malignancies, depletes circulating B cells and can reduce immunoglobulin levels significantly with prolonged use.
Complement deficiencies are rarer but clinically significant. C2 deficiency is the most common classical pathway defect in European-descent populations (~1 in 20,000) and predisposes to encapsulated bacterial infections and systemic lupus erythematosus (SLE). Hereditary angioedema (C1-inhibitor deficiency) affects approximately 1 in 50,000 Australians and requires specific therapies including C1-esterase inhibitor concentrate (Berinert®), icatibant (Firazyr®), and lanadelumab (Takhzyro®).
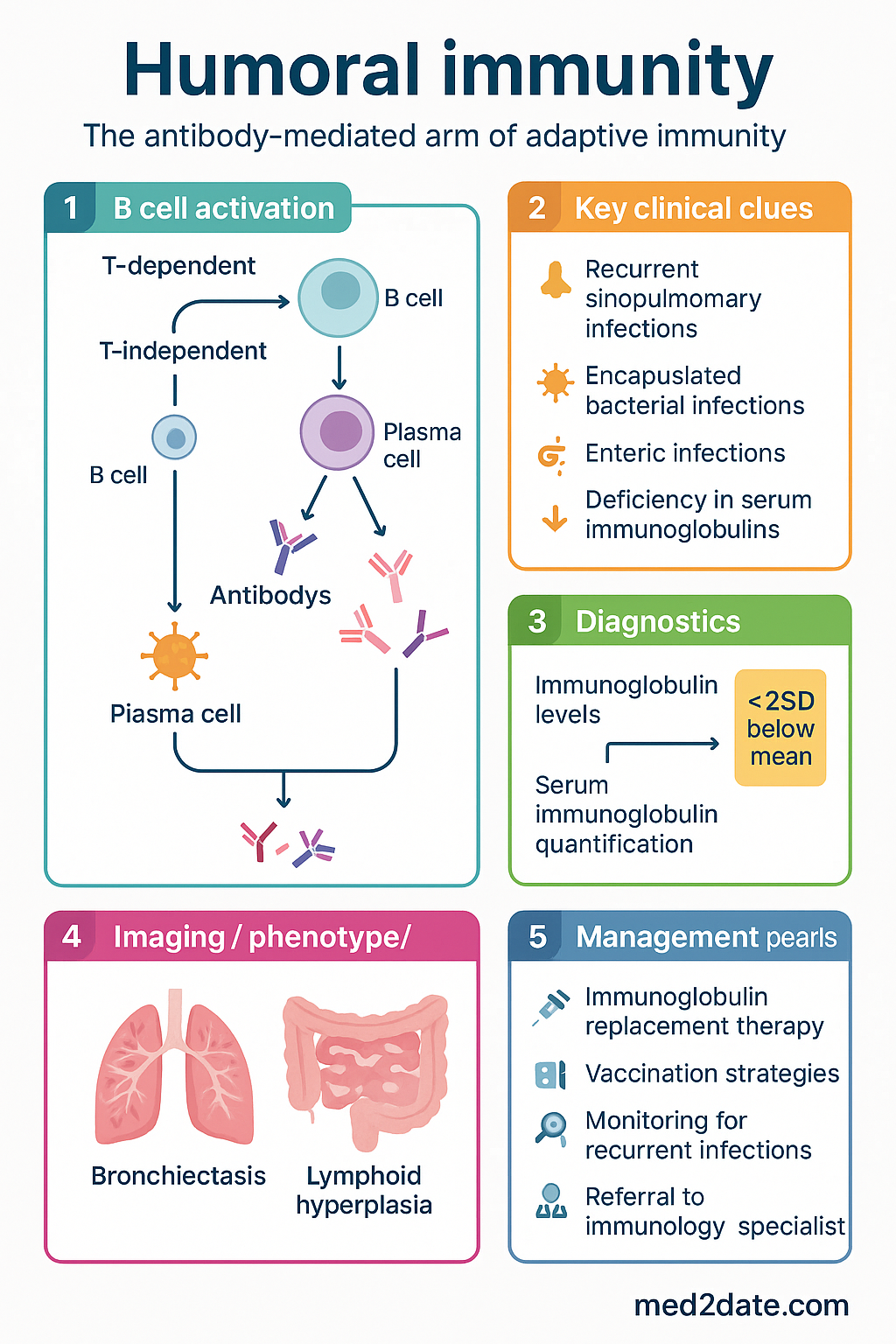
B Cell Activation
B cell activation is the initiating event in the humoral immune response. Naïve B cells reside primarily in secondary lymphoid organs and circulate through blood and lymph. Upon encountering their cognate antigen, B cells become activated, proliferate, and differentiate into antibody-secreting plasma cells or long-lived memory B cells.
T-Dependent B Cell Activation
Most protein antigens require cognate T cell help for full B cell activation — the T-dependent (TD) pathway. This is the dominant mechanism for generating high-affinity antibodies and immunological memory.
T-Independent B Cell Activation
Certain antigens — particularly repetitive polysaccharide structures found on encapsulated bacterial capsules — can activate B cells without T cell help. This T-independent (TI) pathway is subdivided:
- Type 1 TI (TI-1): Polyclonal activators such as bacterial lipopolysaccharide (LPS) that activate B cells via Toll-like receptors (TLR4). At high concentrations, TI-1 antigens activate B cells irrespective of BCR specificity.
- Type 2 TI (TI-2): Highly repetitive epitopes (e.g., pneumococcal polysaccharide capsule) that extensively cross-link BCRs, delivering sufficient intracellular signal without T cell co-stimulation. TI-2 responses predominantly generate IgM (class switching is limited) and produce little immunological memory — this is why conjugate vaccines (e.g., 13vPCV) are superior to plain polysaccharide vaccines (23vPPV) in young children, as they convert the response from TI-2 to TD.
Clinical Relevance of B Cell Activation Defects
Defects at any stage of B cell activation lead to impaired humoral immunity. X-linked agammaglobulinaemia (XLA) results from mutations in BTK (Bruton tyrosine kinase), which is essential for pre-B cell receptor signalling; affected males have virtually absent mature B cells and immunoglobulins. Hyper-IgM syndromes caused by CD40L or CD40 mutations prevent the CD40–CD40L interaction required for germinal centre formation, resulting in markedly elevated IgM with deficient IgG, IgA, and IgE.
Antibody Structure & Function
Antibodies (immunoglobulins) are Y-shaped glycoprotein molecules secreted by plasma cells. Understanding their structure is essential to comprehending their diverse effector functions and the clinical consequences of structural abnormalities.
Basic Structure
Each antibody monomer comprises:
- Two identical heavy (H) chains (~50 kDa each): The heavy chain constant region determines the immunoglobulin class (isotype) — γ (IgG), α (IgA), μ (IgM), δ (IgD), or ε (IgE). Heavy chains contain 3–4 constant domains (CH1–CH4) and one variable domain (VH).
- Two identical light (L) chains (~25 kDa each): Either κ (kappa) or λ (lambda). In humans, the κ:λ ratio is approximately 2:1. Each light chain has one constant (CL) and one variable (VL) domain.
- Disulphide bonds link heavy chains to each other and to light chains. A flexible hinge region between CH1 and CH2 allows the two antigen-binding arms to move independently.
Functional Regions
| Region | Composition | Function |
|---|---|---|
| Fab (Fragment antigen-binding) | VH + CH1 + VL + CL | Antigen recognition and binding. Determines specificity through complementarity-determining regions (CDRs). |
| Fc (Fragment crystallisable) | CH2 + CH3 (+ CH4 in IgM/IgE) | Effector functions: binds Fc receptors (FcγR, FcεR, FcαR) on immune cells; binds C1q to activate complement; mediates placental transfer (IgG via FcRn); determines serum half-life. |
| Hinge region | Between CH1 and CH2 | Flexibility enabling simultaneous binding of two epitopes at varying distances. Susceptible to proteolytic cleavage (papain cleaves above hinge → 2 Fab + Fc; pepsin cleaves below hinge → F(ab′)₂ + pFc′). |
Antibody Effector Functions
Monoclonal Antibodies — Therapeutic Applications in Australia
Knowledge of antibody structure underpins the design of therapeutic monoclonal antibodies (mAbs), many of which are PBS-listed in Australia:
Immunoglobulin Classes
The five immunoglobulin classes (isotypes) differ in structure, distribution, half-life, and effector function. Class-switch recombination (CSR), directed by cytokines from Tfh cells, allows a B cell to change its heavy chain constant region — and therefore its isotype — without altering antigen specificity.
| Isotype | Structure | % Serum Ig | Half-life | Key Functions | Clinical Notes |
|---|---|---|---|---|---|
| IgG | Monomer (150 kDa); 4 subclasses (IgG1–4) | 75–80 % | 21–28 days (IgG1/2/3); 21–28 days (IgG4) | Opsonisation, complement activation (IgG1 > IgG3), ADCC, neonatal passive immunity (placental transfer via FcRn) | IgG1: most abundant subclass; IgG2: important for anti-polysaccharide responses (deficiency → encapsulated infections); IgG3: potent complement activator; IgG4: blocking antibody, poor complement activator |
| IgA | Monomer (serum); dimer with J chain + secretory component (mucosal) | 10–15 % | 5–6 days (serum); varies (secretory) | Mucosal defence; immune exclusion; neutralisation; anti-inflammatory at mucosal surfaces | IgA deficiency (sIgAD): most common primary immunodeficiency (~1 in 500); anti-IgA antibodies in IgA-deficient patients may cause anaphylaxis with blood products containing IgA |
| IgM | Pentamer (900 kDa) with J chain | 5–10 % | 5 days | First-line antibody in primary response; potent classical complement activation; natural antibodies | Pentameric structure provides 10 antigen-binding sites → exceptionally high avidity; cannot cross placenta; elevated in early infection, class-switched memory → IgG |
| IgE | Monomer (190 kDa) | < 0.01 % | 2 days (serum); weeks when bound to FcεRI on mast cells | Type I hypersensitivity (allergic); defence against helminths; mast cell and basophil activation via FcεRI | Lowest serum concentration but highest-affinity Fc receptor (Kd ~10⁻¹⁰ M); basis of allergy and anaphylaxis; targeted by omalizumab |
| IgD | Monomer (185 kDa) | < 1 % | 2.8 days | Co-expressed with IgM on naïve B cells as BCR; role in B cell maturation and innate antimicrobial peptide induction | Function poorly understood; elevated in some chronic infections; IgD myeloma is rare (~1–2 % of myeloma cases) |
IgG Subclasses — Clinical Significance
IgG subclass deficiencies are a recognised cause of recurrent infections in Australian patients, particularly IgG2 deficiency, which impairs responses to polysaccharide antigens. The Australasian Society of Clinical Immunology and Allergy (ASCIA) recommends IgG subclass measurement when total IgG is normal but clinical suspicion of humoral deficiency remains high.
Immunoglobulin Replacement Therapy in Australia
When humoral immunity is severely impaired, replacement with pooled human immunoglobulin is the cornerstone of management. Australian immunoglobulin products are supplied by the National Blood Authority (NBA) under the national blood arrangements.
Complement Activation
The complement system comprises over 30 soluble and membrane-bound proteins that form an enzymatic cascade, bridging innate and adaptive immunity. Complement is activated through three convergent pathways that share a common terminal pathway leading to formation of the membrane attack complex (MAC).
Three Activation Pathways
Terminal Pathway & Membrane Attack Complex
All three pathways converge at C3 cleavage. C3 convertases cleave C3 into C3a (anaphylatoxin) and C3b (opsonin). Addition of C3b to C3 convertases generates C5 convertases (C4b2a3b or C3bBb3b), which cleave C5 into C5a (potent anaphylatoxin and chemotactic factor) and C5b. C5b sequentially recruits C6, C7, C8, and multiple C9 molecules to form the C5b–9 membrane attack complex (MAC), which creates a transmembrane pore causing osmotic lysis of the target cell.
Biological Effects of Complement
| Product | Function | Mechanism |
|---|---|---|
| C3a, C5a | Anaphylatoxins | Mast cell degranulation → histamine release → vasodilation, increased vascular permeability. C5a is the most potent chemoattractant for neutrophils. |
| C3b, iC3b, C3d | Opsonins | Covalently bind pathogen surfaces. Phagocytes recognise C3b via CR1 (CD35) and iC3b via CR3 (CD11b/CD18) and CR4 (CD11c/CD18), enhancing phagocytosis. |
| C3d | B cell co-stimulation | C3d bound to antigen engages CR2 (CD21) on B cells, lowering the activation threshold by up to 1,000-fold — linking complement to humoral immunity. |
| C5b–9 (MAC) | Direct cytolysis | Forms 10 nm pore in target cell membrane → osmotic lysis. Particularly effective against Gram-negative bacteria (thin peptidoglycan wall). |
| C3b-bound immune complexes | Immune complex clearance | Erythrocyte CR1 binds C3b-opsonised immune complexes → transported to liver and spleen → stripped by fixed macrophages. Deficiency → SLE-like immune complex disease. |
Complement Deficiencies & Clinical Syndromes
| Deficiency | Association | Clinical Presentation |
|---|---|---|
| C1q, C2, C4 (early classical) | Systemic lupus erythematosus (SLE) | Immune complex clearance failure → SLE-like disease (C1q deficiency: ~93 % develop SLE); also recurrent encapsulated bacterial infections |
| C3 | Severe recurrent infections | Opsonisation and MAC formation impaired → life-threatening infections with S. pneumoniae, N. meningitidis, H. influenzae; membranoproliferative glomerulonephritis |
| C5–C9 (terminal) | Neisseria infections | MAC formation absent → recurrent invasive meningococcal disease (bacteraemia, meningitis); other infections less affected as opsonisation intact |
| MBL | Increased infection susceptibility (variable) | Common polymorphism (~5 % homozygous deficient); clinically significant mainly in infancy or immunocompromised |
| C1-inhibitor | Hereditary angioedema (HAE) | Uncontrolled contact/kallikrein pathway → bradykinin excess → recurrent episodes of non-pitting subcutaneous/submucosal oedema (face, larynx, GI tract, extremities) |
| Factor H, Factor I | Atypical haemolytic uraemic syndrome (aHUS) | Uncontrolled alternative pathway activation → complement-mediated endothelial injury → thrombotic microangiopathy |
Complement-Targeted Therapies — Available in Australia
Investigations
Laboratory assessment of humoral immunity follows a tiered approach, beginning with widely available screening tests and progressing to specialised assays referred to clinical immunology laboratories.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 10th ed. Philadelphia: Elsevier; 2022.
- 2. Murphy K, Weaver C. Janeway's Immunobiology. 10th ed. New York: Garland Science; 2022.
- 3. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary immunodeficiency (PID) guide for health professionals. Sydney: ASCIA; 2024. Available from: https://www.allergy.org.au/
- 4. Australian Technical Advisory Group on Immunisation (ATAGI). Australian Immunisation Handbook. Australian Government Department of Health; 2024. Available from: https://immunisationhandbook.health.gov.au/
- 5. National Blood Authority (NBA). National Policy on Immunoglobulin Use in Australia. Canberra: NBA; 2023.
- 6. Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205.
- 7. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–1507.
- 8. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797.
- 9. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I — molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262.
- 10. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW; 2023.
- 11. Lucas M, Hugh-Jones K, Welby A, Misbah S, Snowden N, Chapel H. Immunoglobulin replacement therapy for primary immunodeficiency — a practical guide. Clin Exp Immunol. 2010;161(1):1–7.
- 12. Bousfiha A, Moundir A, Tangye SG, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508–1520.
- 13. Maurice PD, Maycock EJ, Whyte IM, Bhagwandeen SB. Complement deficiency and disease in Australian patients. Intern Med J. 2003;33(7):316–320.
- 14. RACGP. Royal Australian College of General Practitioners Red Book: Guidelines for Preventive Activities in General Practice. 10th ed. East Melbourne: RACGP; 2024.