📋 Key Information Summary
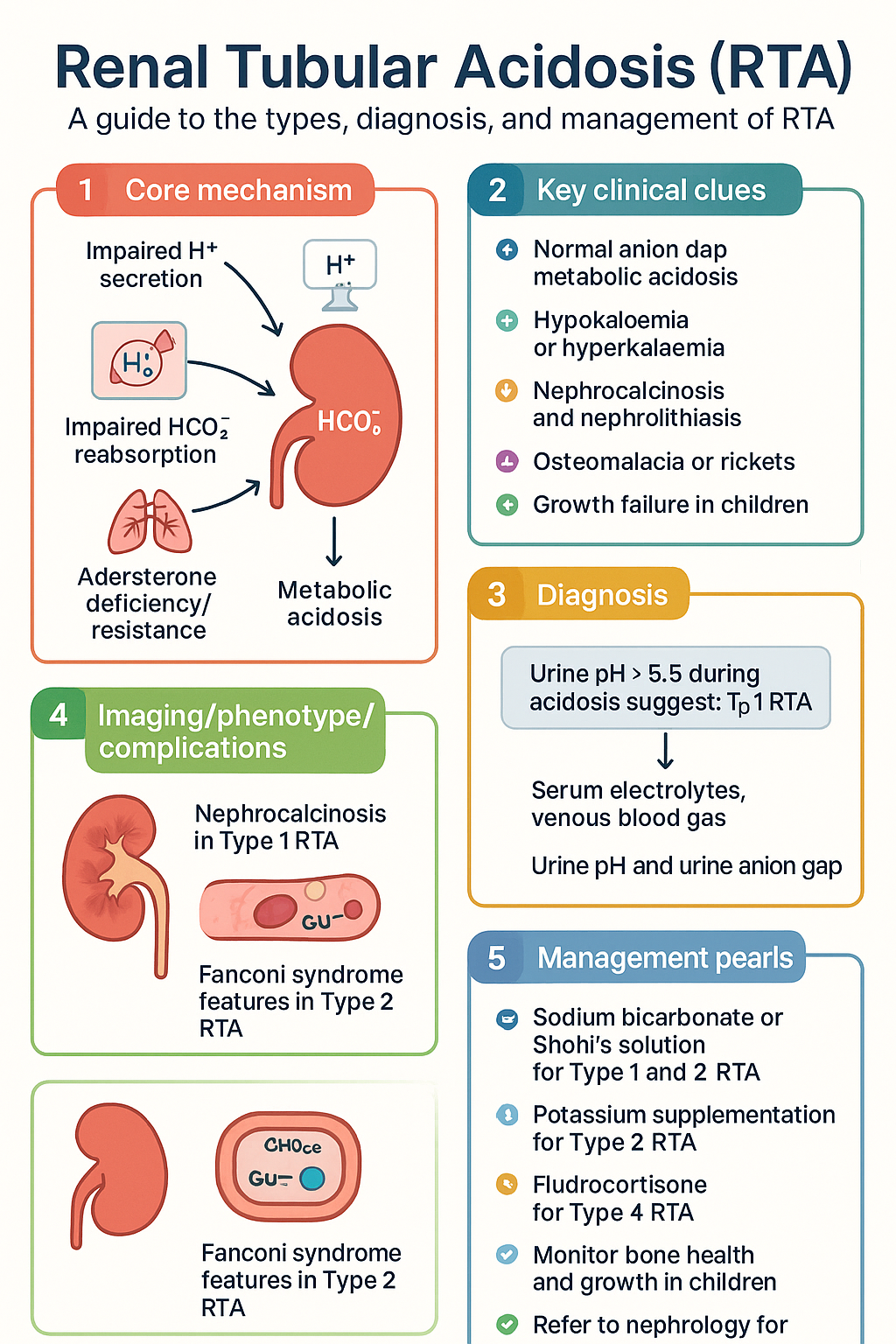
- Renal tubular acidosis (RTA) encompasses Type 1 (distal), Type 2 (proximal), and Type 4 (hyperkalaemic) — all present with normal anion gap metabolic acidosis (NAGMA).
- Type 1 RTA involves impaired H⁺ secretion in the distal nephron; bicarbonate supplementation at 1–2 mmol/kg/day (sodium bicarbonate or Shohl's solution) is first-line therapy.
- Type 2 RTA involves impaired HCO₃⁻ reabsorption in the proximal tubule; higher bicarbonate doses (4–10 mmol/kg/day) are often required, and potassium supplementation is critical.
- Type 4 RTA results from aldosterone deficiency or resistance and is characterised by hyperkalaemia; treatment targets the underlying cause with fludrocortisone or sodium bicarbonate.
- Urine anion gap (UAG) and urine pH help distinguish RTA types: UAG positive with urine pH > 5.5 suggests Type 1; UAG positive with urine pH < 5.5 suggests Type 4; bicarbonaturia on bicarbonate loading suggests Type 2.
- Nephrocalcinosis and recurrent nephrolithiasis are hallmark complications of untreated Type 1 RTA.
- Hypokalaemia is a prominent feature of Type 1 and Type 2 RTA; hyperkalaemia distinguishes Type 4 RTA.
- Causes include autoimmune disease (Sjögren syndrome, SLE), medications (amphotericin B, topiramate, tenofovir), hereditary conditions, and chronic kidney disease.
- Obtain serum electrolytes, venous blood gas, serum calcium, phosphate, alkaline phosphatase, urine pH, and urine electrolytes as baseline investigations.
- Patients with persistent acidosis may develop osteomalacia in adults and rickets in paediatric patients; monitor bone health with DXA and serum alkaline phosphatase.
- Refer to nephrology for diagnostic uncertainty, refractory acidosis, progressive CKD, or suspected inherited RTA syndromes.
- Aboriginal and Torres Strait Islander peoples may have higher prevalence of chronic kidney disease with tubular dysfunction; culturally safe screening and remote-access pathways are essential.
Introduction & Australian Epidemiology
Renal tubular acidosis (RTA) is a heterogeneous group of disorders characterised by normal anion gap metabolic acidosis (NAGMA) resulting from defective renal acid excretion or bicarbonate reabsorption, in the setting of relatively preserved glomerular filtration rate (GFR). Unlike uraemic acidosis, RTA arises from primary tubular dysfunction and cannot be attributed to accumulated organic acids.
Three principal types are recognised: Type 1 (distal RTA), involving a defect in distal tubular H⁺ secretion; Type 2 (proximal RTA), involving impaired proximal HCO₃⁻ reabsorption; and Type 4 (hyperkalaemic RTA), due to aldosterone deficiency or tubular unresponsiveness. Type 3 RTA, a historical hybrid, is rarely used in modern classification.
In Australia, RTA is most commonly encountered secondary to autoimmune conditions such as Sjögren syndrome and systemic lupus erythematosus (SLE), chronic kidney disease, and nephrotoxic medications. Distal RTA has an estimated prevalence of approximately 1 in 10,000 in the general population, though secondary forms are far more common. Aboriginal and Torres Strait Islander peoples carry a disproportionate burden of CKD and its complications, including tubular dysfunction, with rates of kidney disease approximately 2–4 times higher than non-Indigenous Australians (AIHW, 2023).
This guideline provides a structured approach to the diagnosis and management of RTA in the Australian primary care and specialist setting, emphasising evidence-based therapies available under the Pharmaceutical Benefits Scheme (PBS).
Type 1 RTA (Distal) — H⁺ Secretion Defect
Type 1 (distal) RTA results from an inability of the α-intercalated cells of the distal collecting duct to secrete H⁺ into the tubular lumen. This leads to a failure to generate new bicarbonate and maintain systemic acid–base homeostasis, resulting in persistent metabolic acidosis with an inappropriately elevated urine pH (typically > 5.5 even during acidosis).
Aetiology
- Primary (hereditary): Autosomal dominant or recessive mutations in genes encoding the H⁺-ATPase (ATP6V0A4, ATP6V1B1) or the Cl⁻/HCO₃⁻ exchanger (SLC4A1/AE1). Childhood presentation is common in recessive forms; dominant forms may present in adulthood.
- Secondary: Sjögren syndrome (most common secondary cause in Australia), SLE, rheumatoid arthritis, hypergammaglobulinaemia, cryoglobulinaemia, post-renal transplant, medullary sponge kidney.
- Drug-induced: Amphotericin B, ifosfamide, lithium, toluene inhalation, topiramate, zonisamide.
Clinical Features
- NAGMA with serum HCO₃⁻ typically 12–20 mmol/L.
- Hypokalaemia (often severe, K⁺ < 3.0 mmol/L) — due to increased distal Na⁺ delivery and aldosterone response.
- Nephrocalcinosis and nephrolithiasis (calcium phosphate stones) — present in ~70% of hereditary and ~20–30% of secondary cases.
- Osteomalacia (adults) or rickets (children) from chronic buffering of acidosis with bone mineral.
- Growth failure in paediatric patients.
- Proximal muscle weakness and fatigue.
- Urine pH persistently > 5.5 despite systemic acidosis.
Type 2 RTA (Proximal) — HCO₃⁻ Wasting
Type 2 (proximal) RTA results from impaired reabsorption of filtered bicarbonate in the proximal convoluted tubule. Normally, approximately 85% of filtered HCO₃⁻ is reabsorbed proximally. When this mechanism fails, bicarbonate is lost in the urine, leading to NAGMA. The distal nephron retains intact H⁺-secretory capacity, so urine pH can acidify to < 5.5 once plasma bicarbonate falls below the reduced tubular threshold.
Aetiology
- Isolated proximal RTA: Rare; may be hereditary (SLC4A4/NBCe1 mutations, autosomal recessive).
- Generalised proximal tubular dysfunction (Fanconi syndrome): Accompanied by glycosuria, aminoaciduria, phosphaturia, uricosuria, and low-molecular-weight proteinuria.
- Causes of Fanconi syndrome: Tenofovir disoproxil fumarate, ifosfamide, cisplatin, aminoglycosides, valproate, expired tetracyclines, multiple myeloma (light-chain nephropathy), Wilson disease, cystinosis, hereditary fructose intolerance.
Clinical Features
- NAGMA with bicarbonaturia when plasma HCO₃⁻ is above the reduced threshold (typically 14–18 mmol/L).
- Hypokalaemia — exacerbated by alkali therapy, which increases distal sodium delivery and potassium secretion.
- Osteomalacia or rickets (especially in paediatric cases of cystinosis).
- Nephrolithiasis is less common than in Type 1 RTA.
- When Fanconi syndrome is present: glycosuria with normal blood glucose, phosphaturia with hypophosphataemia, aminoaciduria.
Type 4 RTA (Hyperkalaemic) — Aldosterone Deficiency
Type 4 RTA is the most common RTA encountered in clinical practice, particularly in patients with diabetic nephropathy or CKD. It results from either aldosterone deficiency or end-organ resistance to aldosterone in the principal cells of the cortical collecting duct, leading to impaired K⁺ secretion and reduced H⁺ secretion by the α-intercalated cells.
Aetiology
- Aldosterone deficiency: Diabetic nephropathy (hyporeninemic hypoaldosteronism — the most common cause), primary adrenal insufficiency, heparin / low-molecular-weight heparin therapy, NSAIDs, ACE inhibitors, ARBs.
- Aldosterone resistance: Obstructive uropathy, sickle cell nephropathy, pseudohypoaldosteronism type I (autosomal recessive ENaC mutations), amiloride, triamterene, trimethoprim, spironolactone, eplerenone, calcineurin inhibitors (ciclosporin, tacrolimus).
Clinical Features
- Mild NAGMA (HCO₃⁻ typically 18–22 mmol/L).
- Hyperkalaemia (K⁺ > 5.5 mmol/L) — the distinguishing feature.
- The degree of acidosis is often mild, and urine pH can be < 5.5 (distal H⁺ secretion is relatively intact; the primary defect is reduced ammoniagenesis due to hyperkalaemia).
- Often associated with CKD stage 3–4.
- Hypertension (volume expansion from sodium retention).
Pathophysiology
Normal renal acid–base handling involves three integrated mechanisms: proximal reabsorption of ~85% of filtered bicarbonate, distal secretion of H⁺ via H⁺-ATPase and H⁺/K⁺-ATPase, and renal ammoniagenesis (NH₃/NH₄⁺ production from glutamine in the proximal tubule). RTA arises when one or more of these processes are impaired.
| Feature | Type 1 (Distal) | Type 2 (Proximal) | Type 4 (Hyperkalaemic) |
|---|---|---|---|
| Primary defect | Impaired distal H⁺ secretion | Impaired proximal HCO₃⁻ reabsorption | Aldosterone deficiency/resistance |
| Serum HCO₃⁻ | Markedly low (12–20) | Low (14–18) | Mildly low (18–22) |
| Serum K⁺ | Low (hypokalaemia) | Low (hypokalaemia) | High (hyperkalaemia) |
| Urine pH during acidosis | > 5.5 (cannot acidify) | < 5.5 (can acidify below threshold) | < 5.5 (relatively preserved) |
| Nephrolithiasis risk | High | Low | Low |
| Alkali requirement | 1–2 mmol/kg/day | 4–10 mmol/kg/day | Low or none |
In Type 1 RTA, the primary defect results in inability to maximally acidify the urine. In Type 2 RTA, the distal nephron can secrete H⁺, but the excessive proximal bicarbonate loss exceeds distal capacity. In Type 4 RTA, hyperkalaemia itself suppresses proximal ammoniagenesis, contributing to impaired acid excretion — correcting the hyperkalaemia often resolves the acidosis.
Clinical Presentation & Diagnostic Criteria
When to Suspect RTA
- Normal anion gap metabolic acidosis in a patient with normal or near-normal GFR.
- Metabolic acidosis with hypokalaemia (Types 1 and 2) or hyperkalaemia (Type 4).
- Recurrent calcium phosphate kidney stones, particularly in younger patients.
- Osteomalacia or rickets with no clear vitamin D deficiency.
- NAGMA in the setting of Sjögren syndrome, SLE, or multiple myeloma.
- Failure to thrive or growth retardation in children.
Diagnostic Approach
The diagnostic algorithm begins with confirmation of NAGMA and proceeds through urine electrolytes, urine pH assessment, and specialised testing to differentiate RTA subtypes.
Investigations
Diagnosis & Treatment
Type 1 RTA — Treatment
The goal is to normalise serum bicarbonate (target ≥ 22 mmol/L), correct hypokalaemia, and prevent nephrolithiasis and osteomalacia.
Type 2 RTA — Treatment
Higher alkali doses are needed to replace proximal losses. Aggressive potassium supplementation is mandatory as alkali therapy worsens hypokalaemia.
Type 4 RTA — Treatment
Management targets the underlying cause of aldosterone deficiency/resistance and addresses hyperkalaemia.
Monitoring
| Parameter | Frequency | Target |
|---|---|---|
| Serum bicarbonate | Every 1–2 weeks during titration; then every 3 months | 22–26 mmol/L |
| Serum potassium | Every 1–2 weeks initially; then every 3 months | 3.5–5.0 mmol/L |
| eGFR / serum creatinine | Every 3–6 months | Monitor for progression |
| Serum calcium, phosphate, ALP | Every 6 months | Normalise ALP; monitor bone health |
| Renal ultrasound | Annually in Type 1 RTA | Screen for nephrocalcinosis |
| Blood pressure | Every visit | < 130/80 mmHg |
| Urine calcium : creatinine ratio | Every 6 months (Type 1) | < 0.7 mmol/mmol |
Adjust alkali dosing based on serial bicarbonate levels. In patients on fludrocortisone, monitor for oedema, hypertension, and signs of volume overload. Paediatric patients require growth velocity monitoring at each visit.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27(10):3691–3704.
- 2. Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13(8):2171–2177.
- 3. Karet FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol. 2002;13(8):2178–2184.
- 4. Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160–2170.
- 5. Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012;27(12):4273–4287.
- 6. Rennke HG, Denker BM. Renal Pathophysiology: The Essentials. 5th ed. Philadelphia: Wolters Kluwer; 2021.
- 7. Australian Institute of Health and Welfare (AIHW). Chronic kidney disease: Australian facts. Cat. no. PHE 229. Canberra: AIHW; 2023.
- 8. Royal Australian College of General Practitioners (RACGP). National guide to a preventive health assessment for Aboriginal and Torres Strait Islander people. 4th ed. East Melbourne: RACGP; 2024.
- 9. RHDAustralia. Australian guidelines for best practice in the diagnosis and management of acute rheumatic fever and rheumatic heart disease. 3rd ed. Darwin: RHDAustralia; 2020.
- 10. The Royal Australian and New Zealand College of Ophthalmologists (RANZCO) and Kidney Health Australia. Caring for Australasians and New Zealanders with kidney disease. Melbourne: Kidney Health Australia; 2020.
- 11. Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol. 2011;7(2):75–84.
- 12. American Society of Nephrology. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of CKD. Kidney Int. 2024;105(4S):S117–S314.