📋 Key Information Summary
- B–T cell cognate interaction is the central mechanism for high-affinity, class-switched antibody production in germinal centres.
- Antigen-specific recognition requires dual engagement: the B cell receptor (BCR) binds native antigen and internalises it, processing peptides onto MHC class II molecules for presentation to CD4⁺ T follicular helper (TFH) cells.
- The CD40–CD40L axis is the indispensable co-stimulatory signal; CD40L (CD154) on activated T cells engages CD40 on B cells, activating NF-κB, PI3K, and MAPK pathways essential for B cell survival, proliferation, and germinal centre formation.
- Genetic CD40L deficiency causes X-linked Hyper-IgM syndrome (HIGM1) — a severe combined immunodeficiency with absent class switching, recurrent sinopulmonary and opportunistic infections.
- Cytokine signalling from TFH cells (IL-4, IL-21, IL-6, IL-10) directs class switch recombination (CSR) to specific isotypes: IL-4 → IgE/IgG1; IL-21 → IgG1/IgG3; TGF-β → IgA; IFN-γ → IgG2a (mouse) / IgG1-3 (human).
- Class switch recombination (CSR) is an irreversible DNA recombination event at the immunoglobulin heavy-chain constant-region (IgH CH) locus mediated by activation-induced cytidine deaminase (AID).
- AID is also required for somatic hypermutation (SHM); AID deficiency causes Hyper-IgM syndrome type 2 (HIGM2) with elevated IgM, absent IgG/IgA/IgE, and lymphoid hyperplasia.
- Somatic hypermutation in germinal centres introduces point mutations in Ig variable regions; B cells with higher-affinity BCRs are positively selected through competition for T cell help and follicular dendritic cell (FDC) antigen.
- Defective B–T cell interaction underlies multiple primary immunodeficiencies (X-linked agammaglobulinaemia, CVID, HIGM syndromes) and contributes to vaccine failure in immunocompromised patients.
- Therapeutic targeting of CD40–CD40L (e.g., iscalimab/anti-CD40) and cytokine pathways (e.g., dupilumab/anti-IL-4Rα, mepolizumab/anti-IL-5) has transformed management of autoimmune and allergic diseases in Australia.
- Understanding B–T cell co-operation is critical for interpreting vaccine immunogenicity, managing immunodeficiency, and selecting immunomodulatory biologics in Australian clinical practice.
- Aboriginal and Torres Strait Islander populations have higher rates of infectious disease and immune-related conditions; equitable access to immunological diagnostics and biologic therapies remains a national priority.
Introduction & Australian Epidemiology
The adaptive humoral immune response depends on direct cellular interactions between antigen-specific B lymphocytes and CD4⁺ T helper (TH) cells. This co-operation, termed cognate help, occurs primarily in secondary lymphoid organs — lymph nodes, spleen, and mucosa-associated lymphoid tissue (MALT) — and is essential for generating high-affinity, class-switched antibodies, immunological memory, and long-lived plasma cells.
Direct cellular interactions between B cells and T helper cells provide cognate help through CD40L–CD40 co-stimulation and polarised cytokine signalling, enabling antibody class switching and affinity maturation. Without this dialogue, the humoral response remains limited to low-affinity IgM antibodies with poor opsonic and neutralising capacity.
Relevance to Australian Clinical Practice
Disruption of B–T cell co-operation underlies a spectrum of clinical conditions managed in Australian hospitals and primary care:
- Primary immunodeficiencies (PIDs): Australia has a national PID registry managed by the Australasian Society of Clinical Immunology and Allergy (ASCIA). X-linked agammaglobulinaemia (Bruton's, BTK deficiency) affects approximately 1 in 190,000 males; X-linked Hyper-IgM syndrome (CD40L deficiency) affects 1 in 1,000,000 males.
- Common variable immunodeficiency (CVID): The most prevalent symptomatic PID in Australia, with an estimated prevalence of 1 in 25,000. CVID frequently involves defective TFH–B cell interaction, leading to hypogammaglobulinaemia, recurrent bacterial infections, and increased autoimmune and lymphoproliferative complications.
- HIV/AIDS: CD4⁺ T cell depletion by HIV-1 profoundly impairs B cell help, causing hypergammaglobulinaemia paradoxically coexisting with impaired specific antibody responses and vaccine failure. Australia's estimated HIV prevalence is approximately 29,000 (Kirby Institute, 2023), with antiretroviral therapy (ART) restoring but not fully normalising B–T cell co-operation.
- Autoimmune and allergic diseases: Excessive TFH help and dysregulated class switching contribute to pathogenic autoantibodies (systemic lupus erythematosus, rheumatoid arthritis) and IgE-mediated allergy. Australia has among the highest allergy prevalence globally — food allergy affects ~10% of infants, and allergic rhinitis affects ~19% of adults (ASCIA, 2024).
- Vaccine responsiveness: Effective humoral vaccination requires intact B–T cell co-operation. Immunocompromised patients (transplant recipients, rituximab-treated, chemotherapy) frequently have impaired responses to COVID-19, influenza, and pneumococcal vaccines.
- Biologic therapies: Multiple PBS-listed biologics available in Australia target B–T cell interaction pathways, including rituximab (anti-CD20), dupilumab (anti-IL-4Rα), tezepelumab (anti-TSLP), and mepolizumab (anti-IL-5).
Anatomical and Temporal Context of B–T Cell Interaction
Cognate B–T cell interaction occurs in defined microanatomical niches within secondary lymphoid organs:
| Location | B–T Cell Event | Outcome |
|---|---|---|
| T cell–B cell border (lymph node) | Initial cognate interaction; TH priming of naïve B cell | B cell activation, early proliferation |
| Germinal centre (dark zone) | Somatic hypermutation of BCR V regions | Affinity diversification |
| Germinal centre (light zone) | Affinity selection via FDC antigen; TFH rescue signals | High-affinity B cell selection, class switching |
| Extrafollicular foci | T-independent and limited T-dependent responses | Early IgM plasmablasts, short-lived response |
| Mucosal tissues (Peyer's patches, NALT) | TGF-β-driven CSR to IgA; local TFH help | Secretory IgA production |
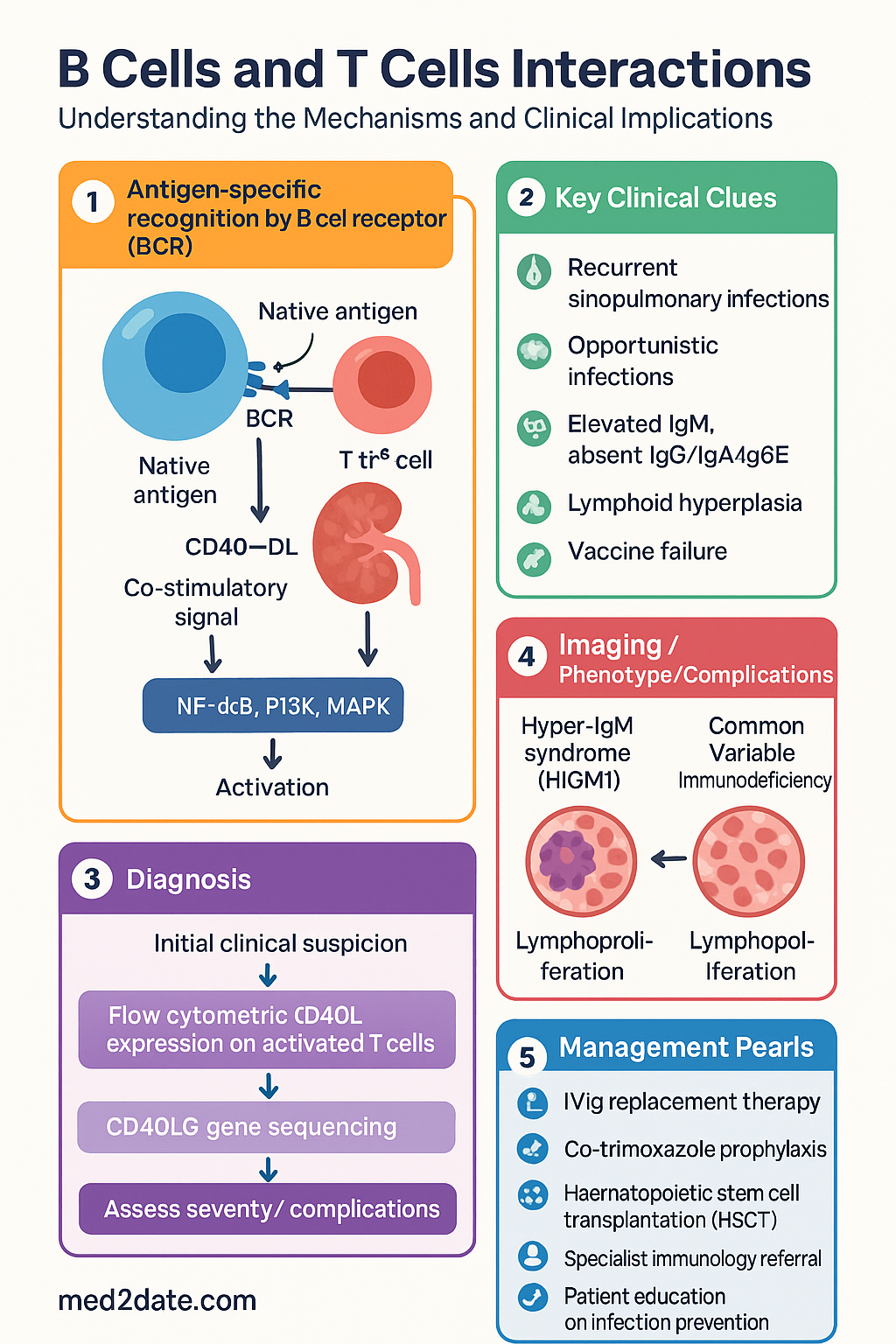
Antigen-Specific Recognition
Antigen-specific recognition is the initiating step of cognate B–T cell interaction. Unlike T cells, which recognise processed linear peptides in the context of MHC molecules, B cells recognise native, three-dimensional conformational epitopes on intact antigens via the B cell receptor (BCR). This dual recognition system creates a requirement for linked recognition — the B cell must internalise the antigen it has bound via the BCR, process it, and present derived peptides on MHC class II molecules to a T helper cell that recognises the same antigen.
B Cell Receptor (BCR) Antigen Engagement
- The BCR consists of a membrane-bound immunoglobulin (mIg) heterodimer (Igα/Igβ, CD79a/CD79b) that transduces activation signals upon antigen binding.
- Naïve mature B cells express surface IgM and IgD; antigen binding triggers BCR crosslinking, phosphorylation of ITAMs on Igα/Igβ by Src-family kinases (Lyn, Fyn, Blk), and recruitment of Syk kinase.
- Downstream signalling cascades — PLCγ2, PI3K, Ras/MAPK, and NF-κB — promote B cell activation, antigen internalisation, and upregulation of co-stimulatory molecules.
- Co-receptor complex (CD19/CD21/CD81) amplifies BCR signalling by 100–1000-fold when antigen is opsonised with complement (C3d). This explains the poor immunogenicity of non-adjuvanted protein antigens and the efficacy of alum-based adjuvants in Australian immunisation schedules.
Antigen Processing and MHC Class II Presentation by B Cells
- Following BCR-mediated endocytosis, antigen is processed in the endosomal/lysosomal compartment into 13–25 amino acid peptide fragments.
- These peptides are loaded onto MHC class II molecules (HLA-DR, HLA-DQ, HLA-DP) in the MHC class II compartment (MIIC) and transported to the B cell surface.
- Activated B cells also upregulate CD80 (B7-1) and CD86 (B7-2), which engage CD28 on the T cell, providing the critical co-stimulatory "second signal" required for full T cell activation.
T Cell Receptor (TCR) Recognition of MHC–Peptide Complex
- The TCR on a CD4⁺ T helper cell recognises the MHC class II–peptide complex presented by the B cell. This interaction is characterised by low affinity (KD ~ 1–100 µM), requiring sustained signal duration (hours) for T cell activation.
- T cell receptor diversity is generated by V(D)J recombination; an estimated 1015–1018 unique TCR specificities are theoretically possible.
- TCR engagement triggers phosphorylation of CD3ζ ITAMs by Lck, recruitment of ZAP-70, and activation of the LAT/SLP-76 scaffold leading to PLCγ1 activation, calcium flux, and NFAT/NF-κB/AP-1 transcription factor activation.
Clinical Significance
- Defects in BCR signalling (Bruton's tyrosine kinase/BTK mutations → X-linked agammaglobulinaemia) cause an absence of mature B cells and profound hypogammaglobulinaemia.
- Defects in MHC class II expression (bare lymphocyte syndrome type II) impair antigen presentation by B cells and other APCs, resulting in severe combined immunodeficiency.
- HLA polymorphism influences peptide presentation efficiency, contributing to inter-individual variation in vaccine responsiveness observed across Australian populations.
CD40–CD40L Interaction
The CD40–CD40L (CD154) axis is the master co-stimulatory pathway for B–T cell cognate interaction. It is essential for germinal centre formation, class switch recombination, somatic hypermutation, affinity maturation, memory B cell generation, and long-lived plasma cell survival. Disruption of this single pathway causes severe immunodeficiency.
Molecular Biology
| Feature | CD40 | CD40L (CD154) |
|---|---|---|
| Expressed on | B cells, dendritic cells, macrophages, endothelial cells, epithelial cells, fibroblasts | Activated CD4⁺ T cells (primarily TFH), platelets, mast cells, basophils, NK cells |
| Molecular structure | Type I transmembrane protein, TNF receptor superfamily (TNFRSF5), 277 amino acids | Type II transmembrane protein, TNF superfamily (TNFSF5), 261 amino acids; homotrimeric |
| Gene | CD40 (chromosome 20q13.12), autosomal | CD40LG (chromosome Xq26.3), X-linked |
| Key signalling adaptors | TRAF1, TRAF2, TRAF3, TRAF5, TRAF6 | TRAF2, TRAF3 (cytoplasmic tail) |
| Downstream pathways | NF-κB (canonical and non-canonical), PI3K/Akt, MAPK (JNK, p38, ERK), PKC | Signals via CD40 on target cell |
Functions of CD40 Signalling in B Cells
- B cell survival: CD40 engagement upregulates anti-apoptotic proteins Bcl-xL and A1/Bfl-1, rescuing B cells from apoptosis in the germinal centre.
- Germinal centre formation: CD40 signalling induces Bcl-6 expression in B cells, which is the master transcription factor for the germinal centre B cell programme.
- Class switch recombination: CD40 signals activate NF-κB, which upregulates AICDA (encoding AID), the enzyme essential for CSR.
- Somatic hypermutation: CD40 co-signals sustain AID expression in germinal centre dark zone B cells, enabling ongoing V region diversification.
- CD80/CD86 upregulation: Enhances the positive feedback loop for T cell co-stimulation.
- Memory B cell differentiation: CD40 signals combined with IL-21 promote memory B cell transcriptional programmes.
- FDC interaction: CD40 signalling enables B cells to interact with follicular dendritic cells bearing immune complexes, a prerequisite for affinity-based selection in the light zone.
CD40 Deficiency (Hyper-IgM Syndrome Type 3, HIGM3)
- Autosomal recessive; clinically indistinguishable from HIGM1 but affects both sexes equally.
- Defective CD40 expression on B cells and monocytes.
- Similar management approach: IVIg replacement, infection prophylaxis, HSCT consideration.
Therapeutic Targeting of CD40–CD40L
- Anti-CD40 antibodies: Iscalimab (CFZ533), a fully human anti-CD40 monoclonal antibody, is in clinical trials for Sjögren's disease, lupus nephritis, and renal transplant rejection in Australian sites.
- Anti-CD40L antibodies: Early trials were complicated by thromboembolism (platelet CD40L engagement). Novel non-FcγR-binding anti-CD40L antibodies (e.g., ruplizumab successors) are under investigation.
- CD40 pathway inhibition represents a promising strategy for transplant immunosuppression and autoimmune disease management, complementing existing biologics available on the PBS.
Cytokine Signalling
Cytokines produced by T follicular helper (TFH) cells and other CD4⁺ T helper subsets provide the critical "third signal" that determines B cell fate — proliferation, class switch recombination destination, plasma cell differentiation, or memory formation. The specific cytokine milieu, combined with CD40 co-stimulation, dictates which immunoglobulin isotype the B cell produces.
Key Cytokines in B–T Cell Co-operation
| Cytokine | Primary Source | Receptor | Effect on B Cells | Class Switch Target |
|---|---|---|---|---|
| IL-4 | TH2 cells, TFH cells | IL-4Rα/γc (Type I) or IL-4Rα/IL-13Rα1 (Type II) | B cell proliferation, survival; CSR to IgG1 and IgE; STAT6 activation → germline ε transcription | IgG1, IgE |
| IL-21 | TFH cells (principal TFH cytokine) | IL-21R/γc | Potent GC B cell proliferation; plasma cell differentiation; STAT3 activation → Blimp-1 upregulation | IgG1, IgG3 |
| IL-6 | TFH cells, FDCs, macrophages | IL-6R/gp130 | Plasma cell differentiation; synergises with IL-21 for antibody secretion | Polyclonal IgG enhancement |
| IL-10 | TFH cells, Treg cells, B cells | IL-10R1/IL-10R2 | B cell proliferation and differentiation to plasma cells; inhibits TH1 cytokines | IgG1, IgG3, IgA |
| IFN-γ | TH1 cells, NK cells | IFNGR1/IFNGR2 | Promotes IgG2a/c class switch (human: IgG1, IgG3); opsonising/complement-fixing isotypes | IgG2a (mouse), IgG1-3 (human) |
| TGF-β | Treg cells, FDCs, epithelial cells | TGF-βRI/RII | CSR to IgA (mucosal immunity) and IgG2b; induction of AID via Smad signalling | IgA, IgG2b |
| BAFF (BLyS) | Macrophages, DCs, FDCs, radiation-resistant stromal cells | BAFFR, TACI, BCMA | B cell survival, maturation; BAFFR deficiency causes hypogammaglobulinaemia | Indirect — supports B cell pool |
| APRIL | Macrophages, DCs, epithelial cells | TACI, BCMA | Long-lived plasma cell survival in bone marrow; IgA CSR at mucosal sites | IgA (TACI-dependent) |
T Follicular Helper (TFH) Cell Biology
TFH cells are the specialised CD4⁺ T cell subset that provides cognate help to B cells within germinal centres. They are defined by:
- Master transcription factor: Bcl-6 (B cell lymphoma 6), which represses alternative T helper programmes (T-bet, GATA-3, RORγt).
- Surface markers: CXCR5 (chemokine receptor for CXCL13, guiding migration into B cell follicles), PD-1 (high), ICOS, CD40L (high), SAP/SH2D1A.
- Key cytokines produced: IL-21 (principal), IL-4, IL-6, IL-10.
- Circulating counterpart: cTFH cells (CXCR5⁺ CD4⁺ T cells in peripheral blood) can be measured as a surrogate for germinal centre activity; elevated in autoimmune diseases (SLE, RA) and useful as a research biomarker.
- SAP (SH2D1A) deficiency causes X-linked lymphoproliferative disease (XLP/Duncan syndrome): impaired TFH–B cell interaction leads to dysgammaglobulinaema and susceptibility to EBV-driven lymphoproliferation. Management includes HSCT at Australian transplant centres.
Cytokine Signalling Pathways — JAK-STAT Axis
Most cytokines relevant to B–T cell co-operation signal through the JAK-STAT pathway:
- IL-4 → JAK1/JAK3 → STAT6 → germline ε promoter activation → IgE CSR
- IL-21 → JAK1/JAK3 → STAT3 (and STAT1) → Blimp-1 (PRDM1) → plasma cell differentiation
- IL-6 → JAK1/JAK2/Tyk2 → STAT3 → acute phase response and plasma cell maturation
- IFN-γ → JAK1/JAK2 → STAT1 → germline γ2a (mouse) promoter → opsonising IgG
- TGF-β → Smad2/3 (non-Smad: MAPK, PI3K) → germline α promoter → IgA CSR
Therapeutic Implications — Biologics Targeting Cytokine Pathways
Class Switch Recombination
Class switch recombination (CSR) is the irreversible DNA recombination event that changes the constant region of the immunoglobulin heavy chain, thereby altering antibody effector function while retaining identical antigen specificity. CSR is entirely dependent on cognate B–T cell interaction — specifically, CD40 co-stimulation combined with cytokine signals.
Molecular Mechanism
- CD40 + cytokine signals converge on AICDA transcription: CD40 engagement activates NF-κB; cytokine (e.g., IL-4) activates STAT6. Both transcription factors bind the AICDA promoter, upregulating AID expression.
- AID deaminates cytosine → uracil in switch (S) region DNA. S regions are repetitive, GC-rich sequences located 5′ of each CH gene (Sµ, Sγ, Sε, Sα).
- Uracil processing generates DNA double-strand breaks (DSBs): Uracil-DNA glycosylase (UNG) removes uracil, creating abasic sites; APE1 (AP endonuclease) cleaves abasic sites. Alternatively, mismatch repair proteins (MSH2/MSH6) recognise U:G mismatches.
- DSBs at the upstream (donor) and downstream (acceptor) S regions are joined by non-homologous end joining (NHEJ), excising the intervening CH genes. The VDJ exon is now juxtaposed to a new CH gene.
- Cytokine determines the acceptor S region: Germline transcription of the target S region (e.g., Sε for IgE) is initiated by cytokine-specific transcription factors, making that S region accessible to AID.
Immunoglobulin Isotype Hierarchy
| Isotype | Subclass | Key Functions | Switch Signal | Serum % (adult) |
|---|---|---|---|---|
| IgM | — | First responder; complement activation (classical pathway); pentameric form | Default (no CSR) | ~10% |
| IgG | IgG1 (60–70%) | Opsonisation, complement fixation, placental transfer (FcRn) | IL-4, IL-21 | ~75% |
| IgG2 (20–30%) | Anti-polysaccharide responses (encapsulated bacteria) | T-independent / IFN-γ | ||
| IgG3 (5–8%) | Potent complement activator, opsonisation; shortest half-life (7 days) | IL-21, IFN-γ | ||
| IgG4 (3–4%) | Anti-inflammatory; blocks IgE-mediated reactions; "blocking antibody" | IL-10 (repeated antigen exposure) | ||
| IgA | IgA1, IgA2 | Mucosal immunity (secretory IgA); immune exclusion; anti-inflammatory | TGF-β, APRIL, BAFF, IL-10 | ~15% serum; predominant mucosal Ab |
| IgE | — | Anti-helminth defence; type I hypersensitivity (allergy); binds FcεRI on mast cells | IL-4 + IL-13 (strong) | <0.01% |
| IgD | — | Co-expressed with IgM on naïve B cells; role in B cell activation and mucosal immunity under investigation | No CSR (co-transcribed with IgM via alternative splicing) | <1% |
Clinical Disorders of Class Switch Recombination
AID and Somatic Hypermutation
- Activation-induced cytidine deaminase (AID) is the single enzyme required for both CSR and SHM.
- In SHM, AID deaminates cytosines in V region DNA; error-prone repair introduces point mutations at a rate ~106× the background genomic mutation rate.
- B cells with higher-affinity BCRs are preferentially rescued by TFH cells (positive selection); low-affinity cells undergo apoptosis (death by neglect).
- This iterative process of mutation and selection — the germinal centre reaction — is the molecular basis of affinity maturation.
- AID overexpression can cause aberrant SHM at non-Ig loci, contributing to B cell lymphomagenesis (e.g., diffuse large B cell lymphoma, Burkitt lymphoma).
Pathophysiology
The pathophysiology of B–T cell interaction disorders can be conceptualised at four levels:
1. Signal 1 Failure — Antigen Recognition
- BTK deficiency (XLA): Absent BCR signalling → B cell developmental arrest at pre-B cell stage → virtually no circulating B cells → agammaglobulinaemia.
- MHC class II deficiency: Absent antigen presentation to CD4⁺ T cells → combined immunodeficiency.
2. Signal 2 Failure — Co-stimulation (CD40–CD40L)
- CD40L deficiency (HIGM1): X-linked. T cells cannot deliver cognate help. No germinal centres form. No CSR or SHM. IgM-only antibodies with poor opsonic function.
- CD40 deficiency (HIGM3): Autosomal recessive. Same phenotype as HIGM1.
- SAP/SH2D1A deficiency (XLP1): TFH cells cannot stabilise conjugation with B cells via SAP-SLAM interactions. Fatal EBV-driven haemophagocytic lymphohistiocytosis.
3. Signal 3 Failure — Cytokine Deficiency
- IL-21R mutations: Recently described; combined immunodeficiency with hypogammaglobulinaemia and impaired T cell function.
- STAT3 deficiency (Hyper-IgE syndrome / Job syndrome): Impaired IL-6 and IL-21 signalling; defective TH17 differentiation; recurrent staphylococcal abscesses, mucocutaneous candidiasis, elevated IgE.
- STAT6 gain-of-function: Causes severe atopic disease with elevated IgE through constitutive IL-4 pathway activation.
4. Effector Failure — AID/UNG/Downstream
- AID deficiency (HIGM2): Autosomal recessive. No CSR or SHM. Lymphoid hyperplasia (massive germinal centres that cannot switch). Elevated IgM, absent IgG/IgA/IgE.
- UNG deficiency (HIGM5): Mild phenotype compared to AID deficiency; residual CSR occurs.
- NHEJ defects (DNA-PKcs, Artemis, Cernunnos/XLF, Ligase IV): Impaired DSB repair affects both CSR and V(D)J recombination → combined immunodeficiency.
Investigations
Investigation of suspected B–T cell interaction defects requires a stepwise approach, from screening immunoglobulin levels to advanced functional and genetic testing. The following investigations are available through Australian laboratories (MBS items indicated where applicable).
Clinical Presentation & Diagnostic Criteria
Clinical Features Suggesting B–T Cell Interaction Defect
B–T cell interaction disorders should be suspected in patients with the following clinical presentations. The Australasian Society of Clinical Immunology and Allergy (ASCIA) provides guidance on PID warning signs.
| Clinical Feature | Suggestive Defect | Age of Onset |
|---|---|---|
| Recurrent sinopulmonary infections (S. pneumoniae, H. influenzae) | XLA, CVID, HIGM, selective IgA deficiency | XLA: 6–12 months; CVID: 2nd–3rd decade; HIGM: infancy |
| Pneumocystis jirovecii pneumonia | HIGM1, HIGM3, SCID | Infancy (often first presentation of HIGM1) |
| Cryptosporidium diarrhoea / sclerosing cholangitis | HIGM1 (CD40L deficiency) | Childhood |
| Chronic/recurrent giardiasis | CVID, selective IgA deficiency | Any age |
| Lymphoproliferation, lymphadenopathy, splenomegaly | HIGM syndromes, CVID, autoimmune lymphoproliferative syndrome (ALPS) | Childhood |
| Neutropenia | HIGM1 (30–50% of patients) | Variable |
| Autoimmune cytopenias, granulomatous disease | CVID | Adolescence/adulthood |
| Family history of PID or early childhood death from infection | X-linked conditions (XLA, HIGM1, XLP) | — |
| Failure to thrive / poor weight gain | Any severe PID | Infancy |
Diagnostic Approach — Australian Framework
Management Principles
Management of B–T cell interaction disorders requires a multidisciplinary approach involving clinical immunology, infectious diseases, haematology, and genetic counselling. Key principles include immunoglobulin replacement, infection prophylaxis, and consideration of definitive therapy.
Immunoglobulin Replacement Therapy
Infection Prophylaxis
- Co-trimoxazole (trimethoprim-sulfamethoxazole): 480 mg PO daily or 960 mg PO 3×/week for Pneumocystis prophylaxis in HIGM syndromes. Paediatric dose: 5–10 mg/kg (trimethoprim component) daily. PBS General Benefit.
- Azithromycin: 250 mg PO three times weekly (or 10 mg/kg weekly in children) for chronic sinopulmonary disease / bronchiectasis prophylaxis. PBS Authority for specific indications.
- Vaccination: Avoid live vaccines in patients with T cell defects or on immunoglobulin replacement. Inactivated vaccines (including COVID-19 mRNA vaccines) are safe but may have reduced immunogenicity. Annual influenza vaccination recommended. Pneumococcal conjugate (13vPCV) and polysaccharide (23vPPV) vaccines per ATAGI/ASCIA guidance.
Definitive Therapy — Haematopoietic Stem Cell Transplantation
- HSCT is curative for HIGM1, HIGM3, and XLP. Australian transplant centres: Royal Children's Hospital Melbourne, Children's Hospital Westmead Sydney, Royal Adelaide Hospital.
- Timing: early transplant (before end-organ damage, e.g., sclerosing cholangitis in HIGM1) improves outcomes.
- Gene therapy for X-linked immunodeficiencies is in clinical trials internationally; not yet available in routine Australian practice as of 2024.
Autoimmune Complications
- CVID patients frequently develop autoimmune cytopenias (ITP, autoimmune haemolytic anaemia) and granulomatous-lymphocytic interstitial lung disease (GLILD).
- Rituximab (anti-CD20, PBS Authority Required for RA — off-label use for CVID autoimmune complications requires specialist management) may be used for refractory autoimmune cytopenias.
- Azathioprine or mycophenolate mofetil as steroid-sparing agents for GLILD (specialist supervision).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
Quick Reference — Key Defects
📚 References
- 1. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132–1148. doi:10.1016/j.immuni.2019.04.011
- 2. Tangye SG, Ma CS, Brink R, et al. The good, the bad and the ugly — TFH cells in human health and disease. Nature Reviews Immunology. 2013;13(6):412–426. doi:10.1038/nri3447
- 3. Stavnezer J, Schrader CE. Ig heavy chain class switch recombination: mechanism and regulation. Journal of Immunology. 2014;193(11):5370–5378. doi:10.4049/jimmunol.1401849
- 4. > Nooman A, Bousfiha A, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. Journal of Clinical Immunology. 2022;42(7):1508–1520. doi:10.1007/s10875-022-01289-3
- 5. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary Immunodeficiency (PID) Guide for Health Professionals. Updated 2024. Available at: https://www.allergy.org.au
- 6. Kirby Institute. HIV, viral hepatitis and sexually transmissible infections in Australia: Annual Surveillance Report 2023. UNSW Sydney.
- 7. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023. Canberra: AIHW.
- 8. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology. 2022;42(7):1473–1507. doi:10.1007/s10875-022-01289-3
- 9. Pharmaceutical Benefits Scheme (PBS). Australian Government Department of Health. Available at: https://www.pbs.gov.au. Accessed 2024.
- 10. Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nature Reviews Immunology. 2013;13(7):519–533. doi:10.1038/nri3466
- 11. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular helper T cells. Annual Review of Immunology. 2016;34:335–368. doi:10.1146/annurev-immunol-041015-055605
- 12. National Health and Medical Research Council (NHMRC). The Australian Immunisation Handbook. Australian Government Department of Health. Updated 2024. Available at: https://immunisationhandbook.health.gov.au