📋 Key Information Summary
- B cell maturation begins in the bone marrow from haematopoietic stem cells and progresses through pro-B → pre-B → immature B cell stages, each defined by rearrangement status of immunoglobulin heavy and light chain genes.
- V(D)J recombination, catalysed by RAG1/RAG2 recombinases, generates the diverse B cell receptor (BCR) repertoire; deficiencies in this process cause severe combined immunodeficiency (SCID).
- Central tolerance in the bone marrow eliminates or edits self-reactive BCRs through receptor editing (secondary light chain rearrangement) and clonal deletion (apoptosis of high-affinity self-reactive clones).
- Peripheral tolerance checkpoints include anergy induction, follicular exclusion, and Fas-mediated deletion — failure of these mechanisms underpins systemic lupus erythematosus and other autoimmune diseases.
- Germinal centre maturation in secondary lymphoid organs drives somatic hypermutation (SHM) and class-switch recombination (CSR), both requiring activation-induced cytidine deaminase (AID).
- T follicular helper (Tfh) cells provide critical CD40L and IL-21 signals to germinal centre B cells, licensing affinity maturation and selection by follicular dendritic cells.
- Plasma cell differentiation is governed by the transcriptional network BLIMP-1 (PRDM1) → XBP-1, which extinguishes PAX5 and BCL6 to permit antibody secretion and survival in bone marrow niches.
- In Australia, primary immunodeficiencies affecting B cell development (XLA, CVID, hyper-IgM syndromes) have a combined prevalence estimated at 1 in 10 000–25 000; ATSI populations may have delayed diagnosis due to limited specialist access in remote areas.
- Flow cytometry immunophenotyping (CD19, CD20, CD10, CD38, CD138, surface immunoglobulin) is the standard investigation and is available through major Australian hospital laboratories (MBS item 65090).
- Therapeutic targeting of B cell lineages — rituximab (anti-CD20), belimumab (anti-BAFF), CAR-T cells, and bispecific antibodies — exploits knowledge of maturation-stage surface antigen expression.
- Immunoglobulin replacement therapy (subcutaneous or IV) is PBS Authority Required for patients with confirmed primary antibody deficiency; monitoring trough IgG every 3–6 months is recommended.
- Distinguishing benign reactive B cell expansions from lymphoproliferative disorders requires integration of morphology, immunophenotyping, and molecular studies (e.g. clonality testing for IGH rearrangement).
Introduction & Australian Epidemiology
B cell maturation encompasses the entire developmental trajectory from multipotent haematopoietic stem cells in the bone marrow through peripheral selection, activation, somatic diversification, and terminal differentiation into antibody-secreting plasma cells or long-lived memory B cells. This process is fundamental to adaptive humoral immunity and forms the biological basis for vaccination, immunodeficiency, autoimmunity, and B cell malignancies.
In Australia, disorders of B cell maturation contribute significantly to clinical practice across multiple specialties. Primary immunodeficiency diseases (PIDs) affecting B cell development — including X-linked agammaglobulinaemia (Bruton disease), common variable immunodeficiency (CVID), and hyper-IgM syndromes — have an estimated combined prevalence of 1 in 10 000 to 1 in 25 000 live births. The Australian Institute of Health and Welfare (AIHW) reports that non-Hodgkin lymphoma, many of which arise from aberrant germinal centre B cell maturation, is the sixth most common cancer nationally, with approximately 6 300 new diagnoses per year.
Understanding normal B cell maturation is essential for interpreting flow cytometry results, diagnosing immunodeficiency, selecting targeted therapies (rituximab, CAR-T, bispecific antibodies), and recognising the pathogenesis of autoimmune and lymphoproliferative conditions. This guideline provides a comprehensive framework of normal B cell maturation with clinical correlates relevant to Australian practice.
Key Terminology
| Term | Definition | Clinical Relevance |
|---|---|---|
| V(D)J recombination | Somatic rearrangement of variable (V), diversity (D), and joining (J) gene segments to generate unique immunoglobulin genes | Defects → SCID (RAG1/2 mutations); errors → B cell lymphomas (translocations) |
| RAG1 / RAG2 | Recombination-activating genes encoding endonucleases essential for V(D)J recombination | Homozygous loss-of-function → T−B−NK+ SCID |
| Somatic hypermutation (SHM) | Introduction of point mutations in immunoglobulin variable regions during germinal centre reaction | AID deficiency → Hyper-IgM syndrome type 2; aberrant SHM → lymphoma |
| Class-switch recombination (CSR) | DNA recombination event that changes the immunoglobulin heavy-chain constant region (e.g. IgM → IgG, IgA, or IgE) | Defects → elevated IgM with low IgG/IgA (hyper-IgM syndromes) |
| AID (AICDA) | Activation-induced cytidine deaminase; enzyme essential for both SHM and CSR | Loss-of-function → Hyper-IgM syndrome type 2 |
| Follicular dendritic cells (FDCs) | Stromal cells in germinal centres that present native antigen on their surface to B cells | Mediate affinity-based selection; long-term antigen retention supports memory |
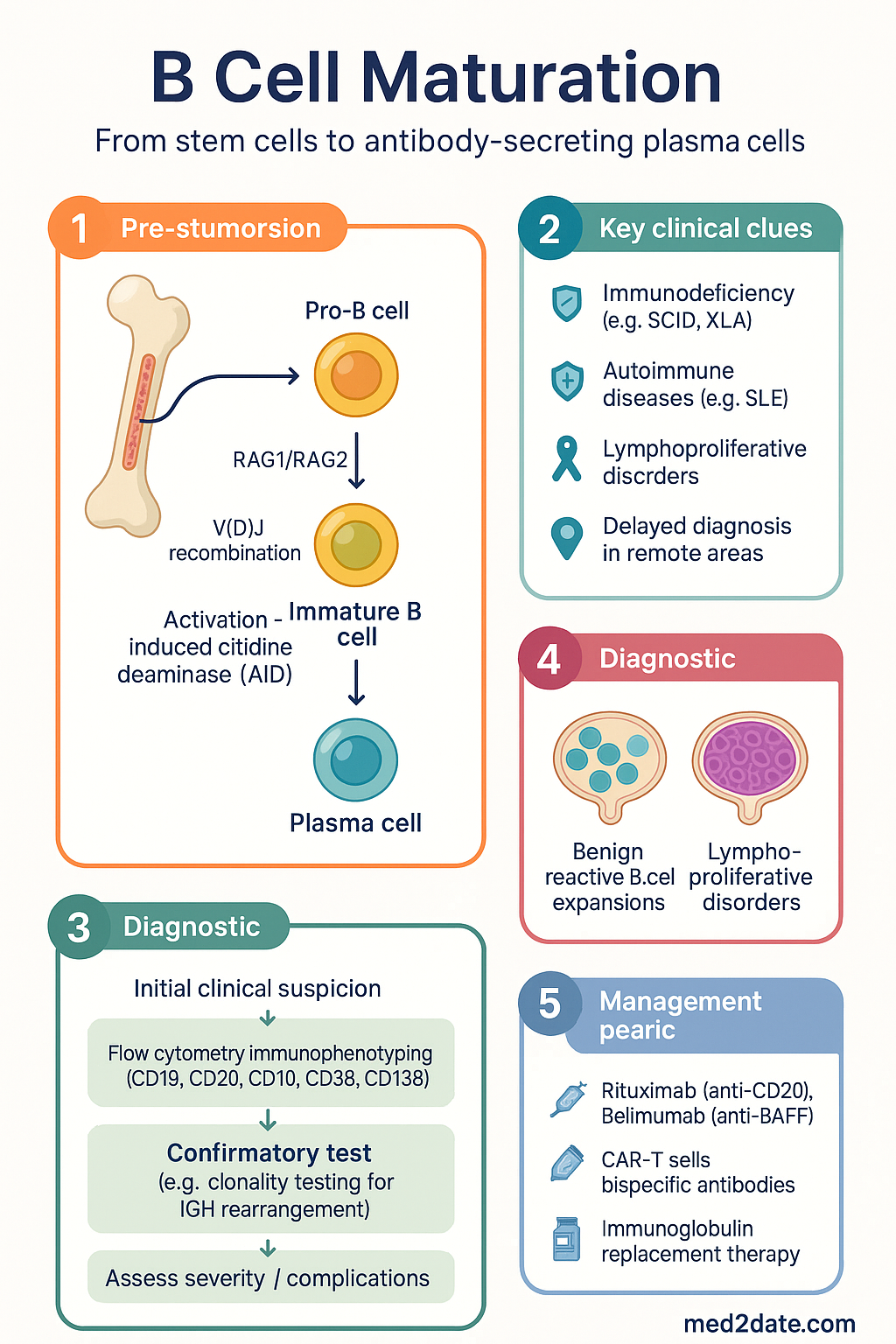
Pro-B to Immature B Cell Development
B lymphopoiesis in humans occurs predominantly in the bone marrow throughout life, though the site of production shifts from foetal liver in utero to bone marrow after birth. The developmental programme from haematopoietic stem cell (HSC) to immature B cell takes approximately 2–3 weeks and is governed by ordered transcription factor expression, cytokine signalling, and the sequential rearrangement of immunoglobulin gene segments.
Developmental Stages & Immunophenotype
| Stage | Surface Markers (CD) | Gene Rearrangement | Key Transcription Factors | Functional Checkpoint |
|---|---|---|---|---|
| Common lymphoid progenitor (CLP) | CD34+, CD38+, CD10+, CD45RA+ | Germline configuration | EBF1, PAX5 (upregulated), E2A | Commitment to B lineage; PAX5 represses alternative lineage genes |
| Early pro-B cell | CD34+, CD10+, CD19+ | D–JH rearrangement on chromosome 14 | PAX5, EBF1, Ikaros | Initiation of heavy chain recombination |
| Late pro-B cell | CD34+/−, CD10+, CD19+ | V–DJH rearrangement | PAX5, FOXO1 | Completion of heavy chain VDJ; surrogate light chain pairing tested |
| Large pre-B cell | CD10+, CD19+, CD34−, cytoplasmic μ heavy chain+ | Heavy chain rearrangement complete; light chain in germline | PAX5, STAT5 (IL-7R signalling) | Pre-BCR checkpoint: μ heavy chain pairs with VpreB/λ5 surrogate light chain → signals proliferation (6–8 cell divisions) |
| Small pre-B cell | CD10+, CD19+, CD34−, surface Ig− | V–Jκ or V–Jλ light chain rearrangement | IRF4, downregulation of FOXO1 | Pre-BCR signals cease; cells exit cycle and rearrange light chain |
| Immature B cell | CD10+, CD19+, CD20lo, surface IgM+, IgD− | Functional BCR (IgM) expressed | PAX5 (maintained), BCL6 (nascent) | Central tolerance checkpoint: cells tested for self-reactivity in bone marrow |
The Pre-BCR Checkpoint
The pre-BCR — comprising a μ heavy chain non-covalently associated with the surrogate light chain (VpreB and λ5/IGLL1) — represents the first major quality-control checkpoint in B cell development. Successful pre-BCR assembly signals the cell to proliferate (clonal expansion of ~6–8 divisions), downregulate RAG1/RAG2, and then exit the cell cycle to commence light chain rearrangement. Failure to produce a functional heavy chain (or surrogate light chain) results in developmental arrest and apoptosis.
V(D)J Recombination — Molecular Detail
V(D)J recombination is initiated by the RAG1/RAG2 recombinase complex, which recognises recombination signal sequences (RSS) flanking each V, D, and J gene segment. RAG introduces a double-strand break at the RSS, and the non-homologous end joining (NHEJ) pathway — involving Ku70/Ku80, DNA-PKcs, Artemis, XRCC4, and DNA ligase IV — resolves the break to create a coding joint (expressed) and a signal joint (excised as a signal circle). This process is:
- Ordered: Heavy chain D–J before V–DJ; heavy chain before light chain; κ before λ
- Stochastic: each allele rearranges independently (allelic exclusion ensures only one specificity per cell)
- Diverse: combinatorial diversity (number of V, D, J segments) plus junctional diversity (P-nucleotides, N-nucleotide additions by TdT, exonuclease trimming) yields >1011 possible specificities
Critical Cytokines & Transcription Factors
- IL-7: The dominant cytokine for early B lymphopoiesis in mice; in humans, IL-7 is important but less exclusively required — human B cell development can partially proceed in IL-7 deficiency, though T cell development is severely impaired.
- SCF (Kit ligand): Supports pro-B cell survival and proliferation via c-Kit (CD117).
- FLT3 ligand: Acts on CLPs and early progenitors.
- PAX5: Master regulator of B lineage commitment; activates B cell–specific genes (CD19, BLNK) and represses alternative lineage programmes (Notch1, M-CSFR). Loss of PAX5 → lineage infidelity.
- EBF1: Cooperates with E2A (TCF3) to activate PAX5 and downstream B cell genes.
- Ikaros (IKZF1): Chromatin remodelling factor required for lymphoid specification; IKZF1 deletions are associated with high-risk B-ALL in paediatric patients.
Central & Peripheral Tolerance
Because V(D)J recombination is a stochastic process, it inevitably generates BCRs that recognise self-antigens. Multiple tolerance checkpoints eliminate or silence these self-reactive clones, both in the bone marrow (central tolerance) and in the periphery (peripheral tolerance). Failure of tolerance is a central mechanism in systemic autoimmune diseases.
Central Tolerance (Bone Marrow)
Immature B cells that express surface IgM are tested against self-antigens in the bone marrow microenvironment. The outcome depends on the affinity and valency of the self-antigen encounter:
Transitional B Cells
Immature B cells that survive central tolerance exit the bone marrow as transitional B cells (T1 and T2 stages in the spleen). These are the final gate before entry into the mature naive B cell pool.
| Stage | Phenotype | Location | Key Features |
|---|---|---|---|
| T1 transitional | IgMhi, IgDlo, CD21lo, CD23−, CD38+ | Splenic red pulp / marginal zone | Highly susceptible to BCR-mediated apoptosis; short-lived (~3 days) |
| T2 transitional | IgMhi, IgDhi, CD21+, CD23+, CD38+ | Splenic follicles | BAFF-receptor (BAFFR/BR3) expression enables BAFF-mediated survival; precursor to mature follicular or marginal zone B cells |
Peripheral Tolerance Mechanisms
Self-reactive B cells that escape central tolerance may encounter self-antigen in the periphery. Multiple mechanisms restrain them:
- Anergy: Chronic low-level BCR engagement without T cell help renders B cells functionally unresponsive. Anergic B cells express reduced surface IgM, fail to signal normally through the BCR, and have shortened half-lives (reduced BAFFR expression).
- Follicular exclusion: Self-reactive B cells are excluded from B cell follicles (and hence from T cell help and germinal centre entry) by competition with non-self-reactive cells for limited BAFF and CXCL13 niches.
- Fas-mediated deletion: If a self-reactive B cell receives BCR and CD40 signals but with inappropriate or absent cytokine support, Fas (CD95) engagement by FasL on T cells induces apoptosis.
- Regulatory B cells (Bregs): A subset of B cells (typically CD19+CD24hiCD38hi or CD19+CD5+CD1dhi) that produce IL-10 and TGF-β, suppressing T cell and dendritic cell activation. Breg deficiency is implicated in SLE, rheumatoid arthritis, and multiple sclerosis.
Clinical Implications of Tolerance Failure
| Tolerance Defect | Mechanism | Associated Condition | Autoantibody Example |
|---|---|---|---|
| Impaired receptor editing | Reduced RAG re-expression or altered chromatin accessibility | SLE | Anti-dsDNA, anti-Smith |
| Excessive BAFF / BAFFR signalling | Rescue of anergic self-reactive B cells | SLE, Sjögren syndrome | Anti-SSA/Ro, anti-SSB/La |
| Defective Fas–FasL pathway | Failure to delete germinal centre autoreactive clones | Autoimmune lymphoproliferative syndrome (ALPS) | Direct Coombs (anti-RBC), anti-phospholipid |
| Breg deficiency | Reduced IL-10–mediated suppression | MS, RA, type 1 diabetes | Variable; disease-specific |
Germinal Centre Maturation
The germinal centre (GC) reaction in secondary lymphoid organs (lymph nodes, spleen, mucosa-associated lymphoid tissue) is the engine of antibody affinity maturation and class-switch recombination. This process underpins effective vaccination and is also the site of origin for the majority of aggressive B cell non-Hodgkin lymphomas in Australia.
Initiation of the Germinal Centre Reaction
The GC reaction is initiated when a mature follicular B cell encounters its cognate antigen presented by subcapsular sinus macrophages or follicular dendritic cells, internalises it via the BCR, processes it, and presents peptide–MHC II complexes to primed CD4+ T cells at the T–B border. This cognate interaction provides three essential signals:
- Signal 1 — BCR engagement: Antigen binding triggers BCR cross-linking and internalisation.
- Signal 2 — CD40/CD40L: CD40L (CD154) on activated T follicular helper (Tfh) cells engages CD40 on B cells — essential for GC formation; CD40 deficiency causes Hyper-IgM syndrome type 3.
- Signal 3 — Cytokines: IL-21 (the dominant GC cytokine), IL-4, and IL-6 drive B cell proliferation, AID expression, and differentiation.
Germinal Centre Architecture — Dark Zone & Light Zone
Dense packing of rapidly dividing B cells (centroblasts) with CXCL12-producing reticular cells. Centroblasts express AID and undergo somatic hypermutation at a rate of ~10−3 mutations per base pair per cell division. Each division introduces 1–2 mutations in the variable region.
Less dense; contains follicular dendritic cells (FDCs) displaying immune complexes, Tfh cells, and non-dividing centrocytes. Centrocytes test their mutated BCR against antigen on FDCs. High-affinity clones capture more antigen, present more peptide–MHC II to Tfh, and receive survival signals (CD40L, IL-21). Low-affinity or self-reactive centrocytes die by apoptosis.
Somatic Hypermutation (SHM)
SHM is initiated by activation-induced cytidine deaminase (AID, encoded by AICDA), which deaminates cytosine to uracil in single-stranded DNA at the immunoglobulin variable region. Error-prone repair by mismatch repair (MMR) and base excision repair (BER, involving UNG and REV1) introduces mutations at and around the original lesion. Key features:
- Targeted to the rearranged V(D)J region (not the constant region)
- Requires transcription through the target locus (RNA pol II–associated cofactors recruit AID)
- Hotspot motifs: WRCY (W = A/T, R = A/G, C, Y = C/T) on the coding strand; complementary motif on the non-coding strand
- Approximately 50% of mutations are synonymous (no change to amino acid); the rest are non-synonymous, creating a diverse pool for selection
Class-Switch Recombination (CSR)
CSR changes the immunoglobulin heavy chain constant region from Cμ (IgM) to Cγ (IgG1–4), Cα (IgA1–2), or Cε (IgE), altering antibody effector function without changing antigen specificity. CSR requires:
- AID: Deaminates cytosines in switch (S) regions upstream of each constant region gene
- UNG and MMR: Process U:A mismatches into double-strand breaks in donor and acceptor S regions
- NHEJ: Joins the broken ends, looping out the intervening DNA as a switch circle
- Cytokine direction: IL-4 → IgE and IgG4; IFN-γ → IgG1 and IgG3; TGF-β → IgA; IL-21 promotes IgG1 and IgG3 in humans
T Follicular Helper (Tfh) Cells — The Orchestration of GC Selection
Tfh cells are specialised CD4+ T cells characterised by expression of CXCR5 (follicular homing), PD-1, ICOS, BCL6 (master transcription factor), and production of IL-21. Their role in the GC includes:
- Affinity selection: Centrocytes that capture more antigen from FDCs present more peptide–MHC II to Tfh, receiving stronger CD40L and IL-21 survival signals.
- Class-switch licensing: Tfh-derived cytokines (IL-4, IL-21) direct CSR.
- Prevention of autoimmunity: Tfh cells that fail to receive adequate co-stimulation become T follicular regulatory (Tfr) cells, suppressing excessive GC reactions.
Clinical correlate — Tfh dysregulation: Excessive Tfh activity is observed in SLE (elevated circulating Tfh, CXCR5+PD-1+CD4+ T cells), Castleman disease (IL-6–driven Tfh expansion), and angioimmunoblastic T cell lymphoma (AITL, a Tfh-derived malignancy). Deficient Tfh function underlies impaired vaccine responses in patients on anti-CD20 therapy (rituximab); vaccination ≥6 months after rituximab cessation is recommended (ASCIA guidelines).
Germinal Centre Output — Memory B Cells vs Plasma Cells
The GC produces two functionally distinct populations:
| Feature | Memory B Cells | Plasma Cells (Short-lived GC-derived) |
|---|---|---|
| Surface markers | IgG/IgA/IgE+, CD27+, CD20+, CD38lo | CD38hi, CD138+, CD20−, surface Iglo/− |
| Function | Rapid recall response upon re-encounter with antigen; can re-enter GC for further maturation | Immediate antibody secretion; migrate to medullary cords / mucosal sites |
| Location | Circulation, marginal zone, mucosal tissues | Lymph node medullary cords, splenic red pulp, bone marrow (if long-lived) |
| Half-life | Decades (self-renewing) | Days to weeks (short-lived); long-lived plasma cells can persist years in bone marrow niches |
| Clinical targeting | Anti-CD20 (rituximab) depletes CD20+ memory B cells but spares plasma cells | Anti-CD38 (daratumumab) and anti-CD138 target plasma cells; proteasome inhibitors (bortezomib) induce apoptosis |
Plasma Cell Differentiation
Plasma cell differentiation is the terminal stage of B cell maturation, transforming a B cell from a signalling lymphocyte into a dedicated antibody-secreting factory capable of producing thousands of immunoglobulin molecules per second. This process is governed by a coordinated transcriptional switch that extinguishes the B cell identity programme and activates the unfolded protein response (UPR) to manage the enormous secretory load.
Transcriptional Regulation of Plasma Cell Differentiation
Short-Lived vs Long-Lived Plasma Cells
| Property | Short-Lived Plasma Cells | Long-Lived Plasma Cells (LLPCs) |
|---|---|---|
| Origin | Extrafollicular response; early GC output | GC-derived; selected high-affinity centrocytes |
| Location | Secondary lymphoid organs, inflamed tissues | Bone marrow survival niches (CXCL12-producing stromal cells, APRIL, IL-6) |
| Lifespan | Days to ~2 weeks | Months to decades |
| Function | Early wave of antigen clearance | Sustained protective serum antibody titres (basis of long-term vaccine protection) |
| Susceptibility to rituximab | Resistant (CD20−) | Resistant (CD20−); explains persistence of protective antibodies post-rituximab in most patients |
| Therapeutic targeting | Corticosteroids, cyclophosphamide | Anti-CD38 (daratumumab, isatuximab), anti-SLAMF7 (elotuzumab), anti-BCMA (belantamab mafodotin), proteasome inhibitors |
Bone Marrow Survival Niches
Long-lived plasma cells home to the bone marrow where they reside in specialised survival niches. These niches provide:
- CXCL12 (SDF-1): Chemokine produced by CXCR4+ stromal cells; mediates homing and retention (CXCR4 antagonism with plerixafor mobilises plasma cells)
- APRIL (TNFSF13): Binds BCMA (TNFRSF17) and TACI on plasma cells → activates NF-κB survival signalling
- IL-6: From mesenchymal stromal cells and osteoclasts → STAT3 activation → MCL-1 and BCL-2 upregulation
- CD44–hyaluronic acid: Adhesion interaction anchoring plasma cells in niches
- Contact with eosinophils: Eosinophils in the bone marrow produce APRIL and IL-6, supporting LLPC survival
Antibody Isotype Functions (Post-CSR Output)
| Isotype | Subclasses | Key Functions | Half-Life (days) |
|---|---|---|---|
| IgM | — | First responder; pentameric form excellent at complement activation (classical pathway) | 5 |
| IgG | IgG1, IgG2, IgG3, IgG4 | IgG1/3: opsonisation, ADCC, complement; IgG2: anti-polysaccharide (encapsulated bacteria); IgG4: anti-inflammatory (blocks rather than activates effector functions) | 21 (IgG1/2/4), 7 (IgG3) |
| IgA | IgA1, IgA2 | Mucosal immunity; secretory IgA (dimeric, J chain + secretory component) neutralises pathogens at mucosal surfaces | 6 |
| IgE | — | Allergic and anti-parasitic responses; binds FcεRI on mast cells/basophils | 2 |
| IgD | — | Co-expressed with IgM on naive B cells; role in B cell activation and upper respiratory mucosal immunity (incompletely understood) | 3 |
Pathophysiology — Disorders of B Cell Maturation
Disruptions at each stage of B cell maturation produce distinct clinical syndromes. Understanding the stage of maturation arrest is critical for diagnosis and targeted management.
Maturation Arrests & Associated Conditions
| Stage of Arrest | Genetic Defect | Condition | Key Laboratory Findings |
|---|---|---|---|
| HSC / CLP | Reticular dysgenesis (AK2) | Severe combined immunodeficiency | Absent T, B, NK cells; neutropenia |
| Pro-B / Pre-B | BTK (XLA), IGHM, IGLL1, CD79a/b, BLNK | Agammaglobulinaemia | CD19+ B cells <1%; all Ig classes profoundly low |
| Immature B cell | PIK3CD (gain-of-function) | Activated PI3Kδ syndrome (APDS) | Elevated transitional B cells; reduced class-switched memory; lymphoproliferation |
| GC reaction — SHM/CSR | AICDA (AID), UNG | Hyper-IgM syndrome types 2 & 5 | Elevated IgM; absent IgG, IgA, IgE; enlarged lymph nodes with GC hyperplasia |
| GC reaction — T cell help | CD40LG, CD40 | Hyper-IgM syndrome types 1 & 3 | Elevated IgM; absent IgG/IgA; neutropenia (CD40LG); opportunistic infections |
| Post-GC / plasma cell | Multifactorial (CVID) | Common variable immunodeficiency | Low IgG + IgA and/or IgM; reduced class-switched memory B cells; diagnosed after age 2 years |
Investigations
Evaluation of B cell maturation disorders requires a tiered approach, beginning with widely available screening tests and progressing to specialised immunological and molecular studies.
Risk Stratification & Severity Scoring
Risk stratification in B cell disorders is guided by the degree of immunodeficiency, presence of end-organ damage, and association with lymphoproliferative or autoimmune complications.
Therapeutic Considerations
Management of B cell disorders is tailored to the underlying maturation defect and its clinical consequences. The following drug cards address the major therapeutic agents targeting B cell lineages in current Australian practice.
Monitoring
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959–971. doi:10.1016/j.jaci.2013.01.046
- 2. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17(5):281–294. doi:10.1038/nri.2017.19
- 3. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2022;40:413–442. doi:10.1146/annurev-immunol-120419-022408
- 4. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15(3):160–171. doi:10.1038/nri3795
- 5. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–1507. doi:10.1007/s10875-022-01289-3
- 6. Australasian Society of Clinical Immunology and Allergy (ASCIA). ASCIA Guidelines for Immunoglobulin Replacement Therapy in Primary Immunodeficiency. Sydney: ASCIA; 2023. Available at: https://www.allergy.org.au
- 7. Slade CA, Bosco JJ, Unglik GA, Bleasel KF, Naglik M, Gaff JM. Deficiency in antibody responses to immunisation in older adults. Clin Exp Immunol. 2023;211(1):1–11. doi:10.1093/cei/uxac115
- 8. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. 2017;17(7):421–436. doi:10.1038/nri.2017.24
- 9. Australian Institute of Health and Welfare (AIHW). Cancer Data in Australia. Canberra: AIHW; 2024. Cat. no. CAN 140. Available at: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia
- 10. Bousfiha A, Moundir A, Tangye SG, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508–1520. doi:10.1007/s10875-022-01352-z
- 11. Rook AH, Bousfiha A, Conley ME, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(9):1400–1412. doi:10.1002/art.40930
- 12. National Health and Medical Research Council (NHMRC). Ethical Conduct in Research with Aboriginal and Torres Strait Islander Peoples and Communities: Guidelines for Researchers and Stakeholders. Canberra: NHMRC; 2018.